
Abstract
Arachis hypogaea is an allotetraploid crop widely grown in the world. Wild relatives of genus Arachis are the rich source of genetic diversity and high levels of resistance to combat pathogens and climate change. The accurate identification and characterization of plant resistance gene, nucleotide binding site leucine rich repeat receptor (NLRs) substantially contribute to the repertoire of resistances and improve production. In the current study, we have studied the evolution of NLR genes in genus Arachis and performed their comparative genomics among four diploids (A. duranensis, A. ipaensis, A. cardenasii, A. stenosperma) and two tetraploid (wild: A. monticola and domesticated: A. hypogaea) species. In total 521, 354, 284, 794, 654, 290 NLR genes were identified from A. cardenasii, A. stenosperma and A. duranensis, A. hypogaea, A. monticola and A. ipaensis respectively. Phylogenetic analysis and classification of NLRs revealed that they belong to 7 subgroups and specific subgroups have expanded in each genome leading towards divergent evolution. Gene gain and loss, duplication assay reveals that wild and domesticated tetraploids species have shown asymmetric expansion of NLRome in both sub-genome (AA and BB). A-subgenome of A. monticola exhibited significant contraction of NLRome while B-subgenome shows expansion and vice versa in case of A. hypogaea probably due to distinct natural and artificial selection pressure. In addition, diploid species A. cardenasii revealed the largest repertoire of NLR genes due to higher frequency of gene duplication and selection pressure. A. cardenasii and A. monticola can be regarded as putative resistance resources for peanut breeding program for introgression of novel resistance genes. Findings of this study also emphasize the application neo-diploids and polyploids due to higher quantitative expression of NLR genes. To the best of our knowledge, this is the first study that studied the effect of domestication and polyploidy on the evolution of NLR genes in genus Arachis to identify genomic resources for improving resistance of polyploid crop with global importance on economy and food security.
Similar content being viewed by others


Introduction
Groundnut or cultivated peanut (Arachis hypogaea) is considered as the most important oil and food legume, grown on 25 million ha with annual production of ~ 46 million tons. A. hypogaea was domesticated in South America ~ 6000 years ago and then widely distributed in post-columbian times1. The genus Arachis consists of 81 species that are mostly diploids. They are classified into nine sections, each with distinct reproductive anatomy, and shows a unique reproductive trait for subterranean fruits2,3,4. Section Arachis is genetically diverse and consist of 30 diploid species and two tetraploids, one wild (A. monticola) and the other domesticated (A. hypogaea)5,6. These tetraploids are the result of interspecific hybridization between two diploid species A. duranensis and (AA, 2n = 20) and A. ipaensis (BB = 2n = 20) that gave rise to wild tetraploids (A. monticola) and after subsequent domestication evolved into a cultivated species A. hypogaea (AABB)1,6,7. Domesticated and repeated cycle of artificial selection have narrowed the genetic base of A. hypogaea which rendered it vulnerable to number of biotic and abiotic stress factors. Peanut crop production is threatened by several disease from bacterial, fungal, virus and nematode diseases including Aspergillus crown rot8, peanut root-knot nematode9 and Cylindrocladium Black Rot (CBR)10 etc. Tapping the wild relatives for broadening the genetic base is an excellent strategy for acquisition of durable resistance against pathogens. Therefore, gaining understanding of underlying molecular mechanism of disease resistance genes, their accurate detection and characterization is vital for achieving higher production rates.
Nucleotide binding site leucine rich repeat receptor (NLRs) recognize the pathogen’s effector via direct or indirect interaction, that activates a number of defensive mechanisms, one of which is hypersensitive response also known as localized programmed cell death11. NLR mainly consist of Nucleotide binding domain (NB-ARC) and C-terminal leucine-rich repeats (NLRs). The NB-ARC domain is the most conserved region to determine the evolutionary relationship between plant NLRs12. There are four major classes of plant NLRs with distinct N-terminal domain fusion: (1) The TIR-NLR subclade containing an N-terminal Toll/interleukin-1 receptor (TIR) domain, (2) CC-NLR subclade containing an N-terminal type Rx-type coiled coil (CC) domain, (3) CCR-NLR subclade containing the RTP8-type CC domain and recently proposed (4) G10 subclade that contains the distinct type of CC and forms a monophyletic group. Previous genome wide analysis on Arachis species were reported in 2003 and 2011 using PCR and BAC library based approach13,14. Genome-wide identification and annotation of NLR genes from plants are challenging owing to their complex sequence diversity and evolutionary history. However, recently released tool NLRtracker identify and characterize NLR genes in high-throughput manner using canonical features of functionally characterized plant resistance genes15.
Genus Arachis provides an excellent opportunity to understand the evolution of NLR genes in diploid and tetraploid species. To this date, comprehensive understanding of evolution of resistance genes in Arachis is not reported. Here we have employed NLRtracker to characterize NLR genes in four diploid and two tetraploid species of Arachis to answer complex questions that are described as follows. What is the effect of allopolyploidy on the NLR evolution in genus Arachis? Whether it causes contraction or expansion of NLRome? Whether this expansion is symmetrical across wild and domesticated tetraploids. What are the suitable wild species that can utilized for introgression into cultivated polyploids to ameliorate in crop production? What are the major evolutionary mechanism employed by Arachis wild relatives to broadens their genetic base for combating biotic and abiotic stress factors?
Methods
Mining of NLR genes in Arachis species
The genome assembly of six Arachis species were downloaded from peanutbase (Table S1). Genome, complete coding sequence (CDS) and reference proteome files for three species A. hypogaea (v.2), A. duranensis (v.1) and A. ipenensis (v.1) were acquired from peanutbase (www.peanutbase.org). Genome assembly of wild relatives A. monticola, A. stenosperma, A. cardenasii were downloaded from NCBI genome portal (Table S1). These three genome were annotated using augustus (v-3.4.0)16 with default settings except for the option of complete gene models (–genemodel = complete). The resulting gff file was parsed into amino acid and coding sequences using two perl scripts (getAnnotFasta.pl and gffread)16. Tetraploids species A. hypogaea and A. monticola were split in to individual A and B genomes in order to simplify the comparison between ancestral species. Reference proteomes from all Arachis species were subjected to the NLRtracker pipeline, which extracts and annotates NLRs from proteins and transcript files. NLRtracker pipeline uses Interproscan17 and predefined NLR motifs18 to extract NLRs and provide domain architecture analyses based on the canonical features found in reference plant NLR genes. NLRtracker annotation of CCR-NLR remained undetermined for this reason, manual curation was performed for each NLR gene using clustering and phylogenetic analysis.
Clusterization and phylogenetic analysis
A library of NB-ARC domain was constructed from reference NLR genes of the PRG database12 and clustered using UCLUST19 with an identity threshold of 50%. The resulting reference genes from each cluster were classified into subgroups already defined by Eunyoung Seo et al.20 and considered as seed probes for phylogenetic and clustering analysis. For comprehensive phylogenetic analysis, extracted NB-ARC domains (output of NLRtracker) from Arachis species were aligned with seed probes of NB-ARC using MUSCLE (version 1.26, Hull, 2009). Subsequent maximum likelihood analysis was performed using IQtree v 2.021, choosing the best-fit model of evolution (-m VT + F + R9) and 1000 bootstrap replicates. We further calculated number of gene cluster and their architecture in each species of genus Arachis by estimating the number of genes in window of 500 kb. Latter we utilized R based conventional script for overlapping visualization.
Chromosomal localization and construction of a syntenic R-gene maps
Coordinates for identified NLR genes were extracted and subjected to density distribution analyses. Unplaced scaffolds were excluded and chromosomal contigs were considered for binning. The number of NLR homologs in 5 Kb bins of each Arachis genome was obtained using “make-windows” and “intersect” commands of bedtools program22. Each bin was then manually labelled with serial number. Using bin number and NLR density value in each bin, linearized version of the genome was visualized using the Rideogram package23. To find the syntenic relationship between NLRs in Arachis species, respective BED files from each species (bin size = 5 kb) were used for the initialization of genomic tracks. BLAST is performed for identification of inter-species genomic similarities, then chromosome and genomic position were retrieved from the GTF file and subsequently sorted according to BLAST output. Genomic linkage was provided on collinearity bases between the genes. The R package “Circlize”24 was used for the visualization of synteny plots.
Evolutionary analysis in Cicer NLRs
Clustalw was used to align each group of paralogs' deduced protein sequences across their respective subgroups (Li 2003). And the obtained alignment was used for a guide in order to align corresponding nucleotide sequences via the usage of the pal2nal software, which is based on the language Perl25. After removing gaps and N-coding codons, ks were estimated using ka/ks calculator under the MA method26. We performed the Fisher test on each paralog selection value, and significant duplication events were kept, and the rest of them were removed (P value > 0.01). Ks-values greater than two (> 2) were eliminated from further consideration since there is a possibility that they suggest substitution saturation. Orthovenn227 was utilized to study orthologs cluster NLR genes. Identified putative NLR genes from each species were queried in locally installed Orthovenn2 program using an E-value of 1e-2 with default settings. All NLR genes identified were subjected to Orthofinder for orthology analysis28. Output containing orthogroups families were labelled manually and species tree was modified into a ultrametric tree using R package APE29. Both files were utilized as input for the CAFE530 and resulting files were manually parsed to evaluate gene gain and loss at each node of species phylogenetic tree. Furthermore, the ortholog sequences between A and B genome of A. hypogaea and A. monticola and their ancestral sequences A. duranensis and A. ipensis were also acquired from Orthofinder28.
RNA-seq based expression analysis
Basal expression level of NLRs identified from this study was evaluated using the available datasets of A. hypogaea and its related species (Table S1). The genotypes utilized different species genus Arachis for expression were different as compared to the genotypes utilized for generating reference genome sequence. First dataset provides comprehensive collection of replicates from pod, seed and shell tissues (PRJNA847769). In the current study, we aligned the raw read sequences using the reference genome of A. hypogaea (v.2) with HISAT31. Alignments were passed to StringTie31 for transcript assembly. Finally, the assembled transcripts and abundance were processed using Ballgown31 for grouping of experimental conditions and determination of differentially expressed between the conditions. In addition, two more datasets were analyzed from project number PRJNA706902, PRNA679430 using similar approach as described earlier. Furthermore, we also evaluated the expression of common NLR genes in progenitor, A. monticola, A. hypogaea and neopolyploids using A. hypogaea genome as reference using PRNA380954 dataset.
Results
Gene mining of NLR genes in Arachis species
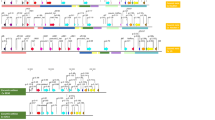
Here we utilized the NLR tracker pipeline15 for NLR genes mining and successive annotations. In case of wild diploid ancestral species for A-genome a total of 521, 354, 284 NLR genes were identified from A. cardenasii, A. stenosperma and A. duranensis. In total, 257 and 454 were identified from A- subgenome of wild (A. monticola) and domesticated (A. hypogaea) tetraploids (AABB) respectively (Fig. 1). Whereas, B-genomes species including A. ipaensis, A. monticola, A. hypogaea contain 290, 397 and 340 NLR genes in their diverse repertoire. Interestingly, A-subgenome from domesticated tetraploid species (A. hypogaea) revealed the significant expansion in the NLR in contrast to A. monticola where reduced number of NLR genes were identified. On the contrary B-subgenome of domesticated tetraploid A. hypogaea revealed contraction as compared to of A. monticola. In addition, among the wild species A. cardenasii shows the expanded NLRome repertoire among all Arachis species (Fig. 1). All four classes of NLR genes were present in all members of Arachis genus. Overall CC-NLR have shown the highest contribution among other classes, on average 51.35% CC-NLR, 35% TIR, 12.1% CCG10 and 1–2% CCR -NLR genes were identified in members of genus Arachis. Interestingly, helper NLR were present in relatively large numbers in tetraploid species especially in A. hypogaea where both AA and BB genome shares 12 CCR-NLR and all the diploid members possess 3–5 CCR except A. ipeansis where 11 CCR helper genes were reported. It is consistent with the previous observation that polyploidization may increase or decrease the number certain genes families32, here CCR shows symmetric expansion in A. hypogaea in both genomes. Distribution of NLR length, length of conserved NB-ARC and species wise domain organization is also provided (Figure S1, S2, S3).

Inverted barplot represent distribution of four classes of NLRs in A. cardenasii, A. stenosperma, A. duranensis, A. monticola, A. ipaensis and A. hypogaea.
Landscape of NLR genes among genus Arachis
We also compared the syntenic relationship between A. hypogaea subgenomes and their progenitor. Highly conserved homeologoues clusters were identified in the syntenic comparison between A-subgenome and B-subgenome of A. hypogaea (Figure S4, A). It should be noted that less syntenic relationships were observed for comparison of each subgenome with its progenitors (Figure S4 B, C). We also studied the landscape of NLR genes in all eight genomes of Arachis genus by plotting the gene density of NLR genes on linearized chromosomes (Fig. 2). Interestingly, A. cardeansii revealed the highest gene density with respect to its size. We also observed the effect of allopolypoid in both wild and domesticated tetraploid species. Interestingly, A-subgenome have shown contraction in wild tetraploid and later on shows significant expansion upon domestication in A. hypogaea. On the contrary, B-subgenome of A. monticola expanded significantly after allopolyploidy with second highest gene density after A. cardenasii (Fig. 2). Overall, synteny and gene density maps strongly suggest that allopolyploidization favors expansion in NLR gene density in Arachis species with the exception of A-subgenome of A. monticola.

Synteny analysis and landscape of NLR genes. (A) Synteny analysis explores depth of evolution and conserved shared synteny between A and B subgenomes of A. hypogaea. (B) The NLR gene density map of all six species of genus Arachis on linearized chromosomes.
In addition we compared the architecture of resistance gene clusters (RGCs) in each species of genus Arachis (Figure S10). Majority of NLR genes were allocated in the form of RGCs. Most of RGCs were allocated on Chr02, Chr04, Chr05, Chr08 and Chr09 (Figure S10). Highest number of total 29 RGCs were found in A. cardenasii and A. stenosperma and the least numbers were observed for A. monticola (A-subgenome). Interestingly, individual number of genes in each cluster were amplified in A. cardenasii, A. hypogea (A-subgenome) and A. monticola (B-subgenome) suggesting active role of tandem duplication in expansion of their NLRome. In addition, presence and absence of RGCs were variable in each genomes.
Phylogenetic analysis and classification of NB encoding genes
Conserved NBARC domain was extracted from each Arachis species and clustered at 75 percent sequence identity using CD-HIT33. Representative members from each cluster (total = 380) were utilized for reconstruction of phylogenetic relationship among A. stenosperma, A. duranensis, A. cardenasii, A. monticola (AA), A. monticola (BB), A. hypogaea (AA), A. hypogaea (BB) and A. ipaensis (Fig. 3). TNL clade was branched out as expected, however TNL remained polyphyletic and three major radiations were observed. On the other hand CNL clade was divided in to three monophyletic major sub-clades CC-NLR, CCR-NLR and CCG10-NLR. CC-NLR was further divided in four major sub-groups CNL-Un, CNL-G11, CNL-G7 and G4. Significant expansion and diversity was observed in G4 and especially in G7 where four strongly supported polyphyletic sub-clades were observed. Interestingly, CNL groups G1, G2, G3, G4, G6, G8 previously identified from Solanaceae family were absent in genus Arachis. That is consistent with the studies from Cicer and dalbergioids, which strongly suggest that Fabaceae members lack G1-G8 groups34,35.

Classification of subgroups of NLR genes using phylogenetic reconstruction. Phylogenetic tree construction is based on the Maximum likelihood method on the VT + F + R9 model. The tree is divided into 7 CNL and 1 TNL subgroups. All the branches are highlighted with their subgroup-specific colors.
Phylogenetic analysis further suggest that progenitor of AA sub-genome, A. duranensis had less number of TIR and CC-NLR genes. After allopolyploidy significant expansion in TIR and CC-NLR genes can be observed. Highest number of these groups can be identified in AA subgenome of domesticated tetraploid A. hypogaea. Interestingly, among all species A. cardenasii has the highest number of TIR and CC-NLR genes considering its diploid nature. This unbalanced gene duplication occurrences across Arachis species suggest possible role of terminal duplication after the divergence from common ancestors.
We also compared the selection pressure within in the pairs of paralogs from four major subgroups (G4, G7, CCG10-NLR, TIR-NLR). G4 (Median = 0.502) and G7 (Median = 0.534) has the highest values of Ka/Ks as compared to other two major groups TIR-NLR (Median = 0.457) and G10 (Median = 0.427), which were evolving under purifying selection. This observation is consistent with the fact that preferential expansion of G4 and G7 is also observed in other Fabacaeae genus Cicer and Dalbergia (unpublished results). In case of A. monticola higher Ka/Ks values of 1.002 was observed for CCG10 subgroup that suggest that its evolving under neutral selection (Figure S5, S6, Table S3).
Duplication assay
Expanded NLRome of A. cardenasii could be because of multiple evolutionary mechanism including duplication, recombination and transposition. Here we explored the duplication history of Arachis NLRs by comparing the Ks values between paralogs of each subgroup. Notably the Arachis lineage have been rapidly accumulating silent changes (~ 1.4 time faster) since the divergence of the Dalbergioid clade1. The closest estimates for divergence between two progenitor of each AA (A. duranensis) and BB (A. ipaensis) sub-genome is recently computed as 2.12 Mya32. However, the precise estimate of divergence of other species from the common ancestor is still not reported. Collective Ks values obtained from all groups suggest one common duplication curve between 0.04 and 0.1 Ks (2.1–6 Mya) (Fig. 4, Table S3). That strongly suggest NLR gene duplication have occurred before the speciation. Highest frequency for gene duplication was observed in A. cardenasii, where peak value of Ks corresponds 0.08 (~ 4.92 Mya). TNL and subgroups G4-CNL, G7-CNL gene had been amplified dramatically through gene duplication events before speciation. Similarly other species A. monticola (B-subgenome) and A. stenosperma also revealed relatively higher frequencies of gene duplication. Interestingly, the progenitor species A. duranensis and A. ipaensis had the least frequency of Ks value for gene pairs. Furthermore, we also tested gene duplication using orthofinder (v 2.5.4: Fig. 5B, D). Consistent with Ks estimates, it suggests that in both A and B genome species highest duplication were observed in the common ancestor of Arachis. Furthermore, Orthofinder provides evidence for relatively higher terminal duplication in A. cardenasii (95) and A. monticola (83: B-subgenome) (Fig. 5B, D). In short, all species represents a common wave of duplication that led to major expansion in NLRome which occurred in the common ancestor of genus Arachis. In addition, terminal duplication was also observed after speciation in specific species that expanded the repertoire of NLR genes in A. cardenasii and A. monticola (B-subgenome).

Duplication history of NLR genes in genus Arachis. Ks-values between paralogs of each subdivision are shown for all six species, where tetraploid species are divided into their constituent subgenomes. (A) X and Y represents the Ks values and frequencies, respectively (B). Overall duplication pattern of NLR genes in genus Arachis.

Ortholog and gene gain and loss analysis. (A, C) Venn diagram represents the shared and common genes (Orthologous clusters) distribution found between A genome related species and B genome related species respectively. (B, D) Gene gain and loss are indicated on each nodes with number of gene gain (green), loss (red) and duplication (blue) for A and B genome related species respectively.
Gene gain and loss
A total of 70 common orthogroups were found conserved in A-genome related species whereas as B-subgenome has 85 common orthogroups (Fig. 5A, C). We constructed the phylogenetic tree for each subgenome with birth and death of genic events among members of genus Arachis. Aeschynomeme evenia was considered as most related outgroup for common ancestor of Arachis. Birth and death model of A-subgenome reveals that contraction of NLR gene families occurred in Aesechynomene evenia which is consistent with overall NLR contraction after whole genome duplication following diploidization. In addition, common ancestor of genus Arachis suggest increased number of gene duplication and gains of 10 additional NLR gene families (Fig. 5B, D). Progenitor of A-subgenome A. duranensis and wild tetraploid A. monticola has shown death of gene families except A. hypogaea where expansion in number of NLR families was observed. Similarly, A. cardenasii revealed the highest expansion of number of diverse gene families probably due to terminal duplication and gained 12 gene families (Fig. 5B, D). Similar trend was found in case of B-subgenome evolution, contraction in the outgroup species and expansion of NLRome in the common ancestor Arachis. Especially in case of A. monticola (B-subgenome) where expansion of NLR genes occurred that is consistent with expansion of other gene families including starch and sugar metabolism, linoleic acid metabolism and cutin synthesis32. In short, asymmetric evolution of NLR genes in A and B sub-genome was observed in wild and domesticated tetraploid species.
Impact of natural and artificial selection pressure on NLR genes
We further studied the impact of natural and artificial selection on NLR gene evolution in both wild and domesticated tetraploid species respectively. For this purpose we compared the ka/ks ratio of orthologs present between subgenomes and their progenitor species (Fig. 6). Ka/Ks values for orthologs between A-subgenome of A. monticola and A. duranensis were significantly higher in A (median = 0.479) as compared B-subgenome (median = 0.455). Similarly, ka/ks values were higher in A (median = 0.488) as compared to B subgenome (median = 0.479) of A. hypogaea (Fig. 6). A bias was observed in selection pressure for A sub-genome NLR genes in both wild and domesticated tetraploid. We also studied the nature of selection pressure on two early diverged species of A-subgenome, for this purpose we compared ka/ks values orthologues of A. cardenasii and A. stenosperma with respect to A. duranensis. These species shows highest degree of natural selection as compared to other wild species, especially A. cardenasii with the selection pressure of M = 0.528. that potentially be the reason for expanded repertoire of NLR genes.

Boxplot represent Ka/Ks values between Arachis species. Middle line between each bare represent median of respective ka/ks.
Expression analysis of NLR genes in Arachis species
We further compared the basal expression level of identified genes in A. hypogaea in three tissue pod, seed and shell. In total 37 NLR genes were constitutively expressed in all three tissues types, notably two genes HV9GRN.1 and 256JRY.1 that belongs to subgroup CCR-NLR and CCG10-NLR respectively shows the highest expression levels in all tissue types (Figure S7). In another study, we evaluated the expression of NLR genes in susceptibe (JL 24) and resistant cultivar (U-475) of A. hypogaea upon Aspergillus flavius infection (Fig. 7). In both cultivars 12 NLR genes were differential expressed and showed strong correlation with disease progresssion. Especially three genes (SMD16A.1, OMH239.1 and WIN0WV.1) revealed higher up-regulation during 3 and 7 dpi in both cultivars (Fig. 8). All three genes are belonged to subgroup G4-CNL which is principal receptor containing coiled coil domain for recogniation of pathogens. Interestingly, no signifcant differences were observed in the expresion profile NLR genes in susceptible and resistant genotype. Presumably other resistance gene including receptor-like kinases (RLK) and receptor-like proteins (RLP) might be responsible for the difference in their genotype.

Comparison of NLR gene expression of susceptible (JL-24) and resistant (U-475) cultivar under Aspergillus flavius infection. Four time points including 1, 2, 3, 7 day post infection (dpi) were selected for the evaluation of their expression.

Comparison of NLR gene expression under drought conditions. Five drought responsive genes were identified indicated in left side.
Recently, it was reported that NLR genes also plays important role under the drought stresss conditions36. Here we tested this hypothesis for A. hypogaea by comparing the expression of NLR genes under well watered versus drought conditions. In this dataset we identified five drought responsive genes (OMH239.1, WIN0ZV.1, UJJ09G5.1, 3L0H24.1 and 84QBSM.1) that were overexpressed during drought conditions. Since this dataset contains biological replicate for 5 days, 7 days and 9 days post drought situation (pds), interestingly we observed highest expression upto 14 fold in 9 days (pds) notably for two genes (0MH239.1, WIN0ZV.1) (Fig. 8).
We also evaluated the expression of NLR genes in both tetraploids and their progenitors as well as synthetic nascent interspecific hybrids and neopolyploids. Bertoili et al.37 reconstructed the hybrids of A. duranensis x A. ipaensis and subsequently induced polyploidy through colchicine treatment. RNA-seq analysis was performed on initial diploids, neopolyploids (1st and 9th generation). Conserved NLR genes that are common to major Arachis species has shown expression bias for interspecific diploid and neo-allopolyploids. Higher individual and cummulative expression levels were observed in synthetic interspecific diploid (AB, A. duranensis x A. ipaensis), neopolyploid (4x) and A. monticola as compared to Arachis hypogaea (Figure S8, S9). In short, NLR genes shows higher quantitative expression levels in A. monticola and neopolyploids (Figure S8).
Discussion
Plants require repertoire of NLR genes for their consistent arm race with the pathogens. Plant genome utilized multiple genetic mechanism for the expansion of NLRome38,39. Tandem duplication is the major driver for their expansion. Secondly the cluster of NLR genes are quite conserved that may cause birth and death of NLR genes via unequal crossing overs or gene conversions38,40. Another mechanism of NLRome expansion in certain plant species is polyploidy, Genus Arachis presents a unique opportunity to understand the evolution of NLR genes, due to presence of diploid progenitor, wild and domesticated tetraploid species. Under normal conditions domestication causes narrowing of genetic base that leads to the loss of important gene involved in biotic and abiotic stress tolerance41. Here we have screened 4 wild diploid and two wild and domesticated tetraploid species to understand the evolution of NLR genes. We employed four major analysis to understand the evolution NLR genes in Arachis that includes gain and loss of NLR genes, their distribution, phylogeny and duplication assay. These analysis strongly suggest that expansion of NLRome in A-subgenome of cultivated A. hypogaea. Similarly, considerable expansion of NLR genes were also observed in B-subgenome of wild tetraploid species A. monticola. In short, our analysis provide basis for asymmetric expansion of NLRome in cultivated and wild tetraploid species. Similar trend were observed in another member species of Fabaceae family, where the allopolyploid T. repens have shown biased expansion of NLRome in A-subgenome42,43.
All diploid wild relatives has shown slightly slower rate of evolution as compared to tetraploid species with the exception of A. cardenasii. This wild species shows the most expanded NLRome in genus Arachis. It has been utilized for the development of disease resilient cultivars in Africa, Asia and Americas. The contribution of this species provide widespread improved food security, environmental and economic benefits41,44. Here in this article we have highlighted the evolutionary mechanism of expanded NLRome. Highest gene duplication frequency with terminal duplication, gene gain and rate of natural selection are the main reason for expanded NLRome. We observed the preferential duplication of subgroup G4 and G7-CNL. Comparative transcriptome analysis of A. cardeansii under infected versus non-infected conditions will allow the identification of effective NLR genes. However, limited dataset were found in databases. Therefore, it is important to generate more genomic and transcriptome resources for the identification of novel resistance genes from A cardenasii.
Reconstructed tetraploid species through Arachis wide crosses can generate spontaneous diversity. Recently generated neo-polyploids from interspecifc hybridization of A. ipaensis versus A. duranensis provides enhanced novelty that can broaden the phenotypic and genotypic plasticity through the mechanism of heterosis and gene redundancy37,45. We tested the expression of NLR genes in different generation of neo diploids and polyploids, our results strongly suggest enhanced qualitative and quantitative expression levels of NLR genes as compared to established polyploids. It might be due to the fact that earlier interspecific hybrids are relatively unstable and shows less regulated expression of certain genes. In addition higher genomic unstable lines tend to perish and only few lineages survives that had stronger mechanism for limiting genomic instability37. Earlier lineages of neo-polyploids can contain novel resistance gene combination that can be introgressed in the cultivated elite lines through conventional and modern approaches.
Polyploidy have played a major role in the expansion of NLRome in genus Arachis. Our results strongly suggest that NLR gene family follows a global trend of asymmetric sub-genome evolution between wild and domesticated tetraploid lineages. It could be due to homeologous sequence exchanges (HSEs) between subgenomes and high frequency of gene duplication. Homoeologous recombination does not only have altered the gene dosage due to chromosomal rearrangement but also results in novel transcript and intergenomic recombinant proteins in nascent allopolyploids5,37. In future HSEs should be studied in detail for understanding the expansion of both nascent and established allopolyploids of Arachis. In addition, structural variation (SVs) also play a pivotal role in the evolution of various gene families across different polyploids e.g. cotton, brassica6,32. In future, we will explore the role of SVs on the evolution of NLR genes and its related families in genus Arachis.
Data availability
Library of identified NLR genes with their comprehensive classification is provided in the supplementary data file. Additional raw and refined output data will be available on request to corresponding author (saad.serfraz@gmail.com).
References
Bertioli, D. J. et al. The genome sequences of Arachis duranensis and Arachis ipaensis, the diploid ancestors of cultivated peanut. Nat. Genet. 48, 438–446 (2016).
Bertioli, D. J. et al. An overview of peanut and its wild relatives. Plant Genet. Resour. Charact. Util. 9, 134–149 (2011).
Grabiele, M., Chalup, L., Robledo, G. & Seijo, G. Genetic and geographic origin of domesticated peanut as evidenced by 5S rDNA and chloroplast DNA sequences. Plant Syst. Evol. 298, 1151–1165 (2012).
Fávero, A. P., Simpson, C. E., Valls, J. F. M. & Vello, N. A. Study of the evolution of cultivated peanut through crossability studies among Arachis ipaënsis, A. duranensis, and A. hypogaea. Crop Sci. 46, 1546–1552 (2006).
Zhang, Z. et al. Homoeologous exchanges occur through intragenic recombination generating novel transcripts and proteins in wheat and other polyploids. Proc. Natl. Acad. Sci. U. S. A. 117, 14561–14571 (2020).
Yin, D. et al. Genome of an allotetraploid wild peanut Arachis monticola: A de novo assembly. Gigascience 7, giy066 (2018).
Zhuang, W. et al. The genome of cultivated peanut provides insight into legume karyotypes, polyploid evolution and crop domestication. Nat. Genet. 51, 865–876 (2019).
Gibson, I. A. S. Crown Rot, a seedling disease of groundnuts caused by Aspergillus niger. Trans. Br. Mycol. Soc. 36, 198-IN3 (1953).
Ballén-Taborda, C. et al. Development and genetic characterization of peanut advanced backcross lines that incorporate root-knot nematode resistance from Arachis stenosperma. Front. Plant Sci. 12, 3078 (2022).
Gai, Y., Deng, Q., Pan, R., Chen, X. & Deng, M. First report of cylindrocladium black rot of peanut caused by Cylindrocladium parasiticum (Teleomorph Calonectria ilicicola) in Jiangxi Province, China. Plant Dis. 96, 586–586 (2012).
Kahraman, A. et al. Distinct subgroups of Cicer echinospermum are associated with hybrid sterility and breakdown in interspecific crosses with cultivated chickpea. Crop Sci. https://doi.org/10.2135/cropsci2017.06.0335 (2017).
Calle García, J. et al. PRGdb 4.0: An updated database dedicated to genes involved in plant disease resistance process. Nucleic Acids Res. https://doi.org/10.1093/nar/gkab1087 (2022).
Ratnaparkhe, M. B. et al. Comparative analysis of peanut NBS-LRR gene clusters suggests evolutionary innovation among duplicated domains and erosion of gene microsynteny. New Phytol. 192, 164–178 (2011).
Bertioli, D. J. et al. A large scale analysis of resistance gene homologues in Arachis. Mol. Genet. Genom. 270, 34–45 (2003).
Kourelis, J., Sakai, T., Adachi, H. & Kamoun, S. RefPlantNLR is a comprehensive collection of experimentally validated plant disease resistance proteins from the NLR family. PLoS Biol. https://doi.org/10.1371/journal.pbio.3001124 (2021).
Stanke, M., Diekhans, M., Baertsch, R. & Haussler, D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics https://doi.org/10.1093/bioinformatics/btn013 (2008).
Jones, P. et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics https://doi.org/10.1093/bioinformatics/btu031 (2014).
Jupe, F. et al. Identification and localisation of the NB-LRR gene family within the potato genome. BMC Genom. 13, 1–14 (2012).
Edgar, R. C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461 (2010).
Seo, E., Kim, S., Yeom, S. I. & Choi, D. Genome-wide comparative analyses reveal the dynamic evolution of nucleotide-binding leucine-rich repeat gene family among solanaceae plants. Front. Plant Sci. 7, 1205 (2016).
Nguyen, L. T., Schmidt, H. A., Von Haeseler, A. & Minh, B. Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274 (2015).
Quinlan, A. R. & Hall, I. M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics https://doi.org/10.1093/bioinformatics/btq033 (2010).
Hao, Z. et al. RIdeogram: Drawing SVG graphics to visualize and map genome-wide data on the idiograms. PeerJ Comput. Sci. https://doi.org/10.7717/peerj-cs.251 (2020).
Gu, Z., Gu, L., Eils, R., Schlesner, M. & Brors, B. Circlize implements and enhances circular visualization in R. Bioinformatics https://doi.org/10.1093/bioinformatics/btu393 (2014).
Suyama, M., Torrents, D. & Bork, P. PAL2NAL: Robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. https://doi.org/10.1093/nar/gkl315 (2006).
Zhang, Z. KaKs_calculator 3.0: Calculating selective pressure on coding and non-coding sequences. Genom. Proteom. Bioinform.s https://doi.org/10.1016/j.gpb.2021.12.002 (2022).
Xu, L. et al. OrthoVenn2: A web server for whole-genome comparison and annotation of orthologous clusters across multiple species. Nucleic Acids Res. https://doi.org/10.1093/nar/gkz333 (2019).
Emms, D. M. & Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 20, 1–14 (2019).
Paradis, E. et al. ape: Analyses of PHYLOGENETICS AND EVOLUTIon. Bioinformatics 20, 289–290 (2014).
Mendes, F. K., Vanderpool, D., Fulton, B. & Hahn, M. W. CAFE 5 models variation in evolutionary rates among gene families. Bioinformatics 36, 5516–5518 (2020).
Pertea, M., Kim, D., Pertea, G. M., Leek, J. T. & Salzberg, S. L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 11, 1650–1667 (2016).
Yin, D. et al. Comparison of Arachis monticola with diploid and cultivated tetraploid genomes reveals asymmetric subgenome evolution and improvement of peanut. Adv. Sci. 7, 1901672 (2020).
Fu, L., Niu, B., Zhu, Z., Wu, S. & Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152 (2012).
Tariq, T., Haider, Z., Rani, S. & Serfraz, S. Evolution of NLR genes in Cicer species revealed a dynamic expansion of the NLRome in C. echinospermum. Unpubl. articl (2022).
Rani, S., Tariq, T., Haider, Z. & Serfraz, S. Evolution of NLRome in Dalbergioids. Unpubl. Artic. (2022).
Irulappan, V. et al. Drought stress exacerbates fungal colonization and endodermal invasion and dampens defense responses to increase dry root rot in chickpea. Mol. Plant-Microbe Interact. 35, 583–591 (2022).
Leal-Bertioli, S. C. M. et al. Spontaneous generation of diversity in Arachis neopolyploids (Arachis ipaënsis x Arachis duranensis)4x replays the early stages of peanut evolution. Genes Genomes Genet. 11, 289 (2021).
Mizuno, H. et al. Evolutionary dynamics and impacts of chromosome regions carrying R-gene clusters in rice. Sci. Rep. 10, 872 (2020).
Wu, J. Y., Xue, J. Y. & Van de Peer, Y. Evolution of NLR resistance genes in magnoliids: Dramatic expansions of CNLs and multiple losses of TNLs. Front. Plant Sci. 12, 2998 (2021).
Chen, J. Y. et al. Genome-wide analysis of the gene families of resistance gene analogues in cotton and their response to Verticillium wilt. BMC Plant Biol. 15, 1–15 (2015).
Bertioli, D. J. et al. Legacy genetics of Arachis cardenasii in the peanut crop shows the profound benefits of international seed exchange. Proc. Natl. Acad. Sci. U. S. A. 118, e2104899118 (2021).
Griffiths, A. G. et al. Breaking free: The genomics of allopolyploidy-facilitated niche expansion in white clover. Plant Cell 31, 1466–1487 (2019).
Areej, A. et al. Investigation of NLR genes reveals divergent evolution on NLRome in diploid and polyploid species in genus trifolium. Genes 14, 867 (2023).
Stalker, H. T. Utilizing wild species for peanut improvement. Crop Sci. 57, 1102–1120 (2017).
Nguepjop, J. R. et al. Evidence of genomic exchanges between homeologous chromosomes in a cross of peanut with newly synthetized allotetraploid hybrids. Front. Plant Sci. 7, 1635 (2016).
Acknowledgements
This work was funded by Researchers Supporting Project Number (RSP2023R26), King Saud University, Riyadh, Saudi Arabia.
Author information
Authors and Affiliations
Contributions
S.S. conceived the study, performed analyses and analyzed the data with help from M.R., S.R., Z.H., S.S., A.B., M.D., F.S., M.S.N.R., M.M., V.S., S.A., H.M.R. and A.S. drafted the manuscript and all authors contributed to the final version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rizwan, M., Haider, S.Z., Bakar, A. et al. Evolution of NLR genes in genus Arachis reveals asymmetric expansion of NLRome in wild and domesticated tetraploid species. Sci Rep 13, 9305 (2023). https://doi.org/10.1038/s41598-023-36302-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-36302-1
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}