









-
Asiatic hybrid lily (Lilium spp.) is one of the most important cultivars of Lilium, mainly distributed in East Asia, and is derived from interspecific crosses of the Sinomartagon section[1]. Asiatic hybrids are widely cultivated for landscaping, and it has high ornamental value due to its elegant floral shape and brilliant colors. The domestication of the double-flower is of great value in ornamental plants. In Lilium, the origin type of double-flower is mainly pistil or stamen petalization. Double-flower lily cultivars are favored for their layered flowers and the abortive stamens with no pollen. However, most double-flower cultivars are concentrated in Oriental hybrids. The Asiatic hybrid 'Annemarie's Dream' is a double-flowered cultivar whose stamens convert into petaloid organs in the 3rd whorl in varying degrees, and the other stamens become pollen-aborted degenerate stamens (Fig. 1). Therefore, this unique feature provides the materials for studying the stamen petaloid during the double-flower development and thus offer a basis for double-flower breeding by altering flora phenotype.

Figure 1.
Floral phenotype of the Asiatic hybrid lily 'Annemarie's Dream'. (a) S1: bud stage. (b) S2: full-bloom stage. Bar = 1 cm.
Owing to its important role in both plant reproduction and ornamental value, floral organ identity has been well studied in Arabidopsis and Antirrhinum. Bowman et al. proposed that flora organ identity is regulated by the ABC model[2,3]. The flower organs of angiosperms are generally composed of four classes in the whorl, the first floral whorl are the sepals, the second whorl are the petals, the third whorl are stamens and the fourth whorl are carpels[4]. Based on the functional verification of double or triple mutants of flower development[5], each floral organ development is controlled by five types of homeotic genes in the MADS-box gene family, A, B, C, D, and E, individually or in combination. The ABC model confirmed that the A-class genes including APETALA1 (AP1), APETALA2 (AP2) determines the formation of sepals in the first whorl, the combined expression of B-class genes APETALA3 (AP3), PISTILLATA (PI) and A-class genes specify the development of petals in the second whorl, with third whorl stamens being specifically controlled by B and C-class genes AGAMOUS (AG), and the C-class gene acts to form carpels in the fourth whorl[6,7]. In addition, the D-class genes SEEDSTICK (STK) and SHATTERPROOF (SHP) determine the formation of carpels[8]. Subsequent research found E-class genes SEPALLATA (SEP) can interact with other class genes and enable the normal development of four whorls of floral organs[9]. As research has advanced, a floral quartet model has been proposed, which suggests that identity of floral organs is regulated by tetrameric protein complexes of MIKC-type MADS-domain transcription factors A-, B-, C- and E-class of the MADS-box family in Arabidopsis[10]. Petal development is regulated by the interaction of genes in B-class (AP3/PI) with A-class (AP1) and E-class (SEP), while the stamen is specifically controlled by B-class genes in combination with those in E-class and C-class (AG)[11].
Currently, the research on the regulation of the double-flower formation mechanism is mainly focused on the C-class AGAMOUS ortholog gene. Loss of function of the C-class AGAMOUS gene in Arabidopsis will change the A-class gene expression boundary, from the first and second whorls to the third whorl stamens[12,13]. In some ornamental plants, loss of function or reduced expression of the AG gene will convert stamens into petals[14−16]. Ma et al. identified and isolated the homologous AG genes in single-flowered and double-flowered varieties in Kerria japonica, and ectopic expression analysis in Arabidopsis showed that the AG gene of double-flowered flowers do not display the function of the C-class[17]. In addition, François et al. identified two alleles of the A-class gene AP2 in a rose double-flowered mutant, one of the alleles contains a transposon with an insert intron that produces an AP2 mutant with miR172 resistance to regulate the expression of the AG gene to control the formation of double-flowered flowers[18]. In Lilium, an AGAMOUS-like gene was isolated from 'Elodie', which is correlated with the degree of petaloidy of the stamens[16]. Meanwhile, it has been shown that some genes involved in phytohormone signaling transduction and TFs regulation can also work with MADS-box transcription factors to control the development of plant flowers[19,20]. Transcriptome analysis during flower development revealed that MADS-box and hormone signal transduction related genes, play a vital role in the stamen petaloid in Lagerstroemia speciosa and Nelumbo nucifera[21,22].
The above-mentioned research results indicate that the morphology of stamen petaloid may be regulated by the 'ABCE' model and phytohormone-related genes during double-flower development. However, it is unclear whether the molecular regulatory mechanism of stamen petaloid during flower development is consistent with the 'ABCE' model in Asiatic lily hybrids. In the present study, we conducted RNA-seq to comparatively analyse the transcriptomic differences during the homeotic transformation of stamen into tepal at two floral development stages (i.e., stamen, petaloid stamen and inner tepal). The enrichment analysis of key DEGs indicated that plant hormone signal transduction pathways are strongly involved in stamen petaloid. In addition, the expression patterns of these genes were analyzed in this transition. With the help of weight gene co-expression network analysis (WGCNA), our results provided insight into the molecular regulatory mechanism regulating stamen petaloid. In conclusion, we hypothesized that the significant down-regulation of CL14315.Contig2_All (LiAG), CL3014.Contig2_All (PYL) and CL5627.Contig1_All (GID2) may lead to stamen petaloid in Asiatic hybrids 'Annemarie's Dream'.
-
The floral organ shape of ornamental plants is an important factor that determines the ornamental value. Normally, ornamental plants generally have four whorls of floral organs, which are sepals, petals, stamens and carpels. In lilies, sepals and petals are collectively referred to as the tepals. We divided the Asiatic hybrid lily 'Annemarie's Dream' flower development into two periods as S1 (bud stage) and S2 (full-bloom stage) (Fig. 1a & b). The double-flowered lily 'Annemarie's Dream' have three outer tepals and three inner tepals in whorls one and two, petaloid stamen and normal stamen in whorl three, and one pistil in whorl four. In the S1 stage, we discovered that stamens are converted to petaloid organs in varying degrees and the other stamens become pollen-aborted degenerate stamens. The normal tepal, staminode, petaloid stamen and pistil continue to elongate, and the flowering process is completed at the S2 stage. Therefore, 'Annemarie's Dream' provides a good model for research on the molecular mechanism of stamen petaloid formation during double-flower development.
Transcriptome sequencing and de novo assembly
-
To further explore the molecular mechanism underlying stamen petaloid phenotype during double-flower development, 18 cDNA libraries at two double-flower development stages were sequenced using a Illumina Hiseq platform. Comparative transcriptomic analysis among bud stage stamen (Budst), bud stage petaloid stamen (Budpest), inner bud tepal (Inbud), full-bloom stage stamen (ST), full-bloom stage petaloid stamen (PEST) and full-bloom stage inner tepal (InTE) were conducted. A total of 185.94 Gb clean data were generated from 18 cDNA libraries, with an average of 10.33 Gb of clean data per sample for further analysis (Supplemental Table S1). After de novo assembly of clean reads using Trinity, 190,488 unigenes were obtained with total length, average length, N50 and GC content of 147,192,422 bp, 772 bp, 1,438 bp and 44.34%, respectively. In addition, seven databases (KEGG, GO, NR, NT, SwissProt, Pfam and KOG) were used to annotate all unigenes to provide gene function information. In total, 97,282 (51.07%) of 190,488 unigenes were annotated. Within these databases, 87,454 genes (45.91%) were annotated in Nr; 57,964 genes (30.43%) in NT; 65,424 genes (34.35%) in SwissProt; 68,882 genes (36.16%) in KOG; 67,991 genes (35.69%) in KEGG; 28,443 genes (14.93%) in GO and 60,776 (31.91%) in Pfam.
Analysis of differentially expression genes (DEGs) among floral organs
-
To investigate the transcriptional differences associated with stamen petaloid in double- flowers, we conducted pairwise comparison among inner tepal, stamen and petaloid stamen at two development stages (Fig. 2). In total, 37,549 DEGs were identified at a fold change ≥ 2 and adjusted P-value ≤ 0.001, among which the highest number of DEGs was ST vs PEST with 58,887. In some comparisons (ST vs PEST, ST vs InTE), the down-regulated DEGs were detected more than up-regulated DEGs, and this trend was most notable at the full-bloom stage (Fig. 2a). We determined significant DEGs between ST vs PEST, ST vs InTE, InTE vs PEST at full-bloom stage and Budst vs Budpest, Inbud vs Budpest, Budst vs Inbud at the bud stage by estimating the gene expression level using FPKM, with a log2 fold change greater than 2 and Q-value < 0.001. We found that there were 41,683, 41,343 and 25,824 DEGs between ST vs PEST, ST vs InTE and InTE vs PEST respectively (Fig. 2a). The number of overlapping DEGs detected in ST vs PEST and ST vs InTE but not in InTE vs PEST was 24,057 (Fig. 2b), which might be related to the formation of stamen petaloid. In the comparison of bud stage, there were 24,878, 21,136 and 29,460 DEGs between Budst vs Budpest, Inbud vs Budpest and Budst vs Inbud respectively (Fig. 2c). The results were similar to those in the blooming stage, with a higher number of DEGs for the comparison of stamens vs petaloid stamens/inner bud tepal. Additionally, the largest number of DEGs in the ST vs Budst comparison was 42,059 accompanying the flowering process (Fig. 2d).

Figure 2.
Analyses of DEGs at two development stages of stamen, petaloid stamen and inner tepal. (a) The number of up- and downregulated DEGs in 9 comparisons. (b) Venn diagram of DEGs in ST vs PEST, ST vs InTE and InTE vs PEST. (c) Venn diagram of DEGs in Budst vs Budpest, Budst vs Inbud and Inbud vs Budpest. (d) Venn diagram of DEGs in ST vs Budst, PEST vs Budpest and InTE vs Inbud. (e) KEGG pathway enrichment analysis of DEGs for ST vs PEST. (f) KEGG pathway enrichment analysis of DEGs for ST vs InTE.
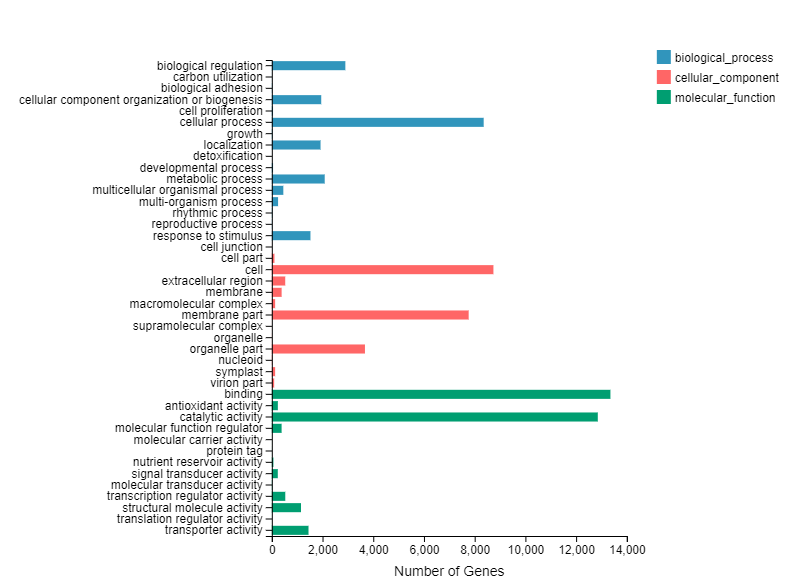
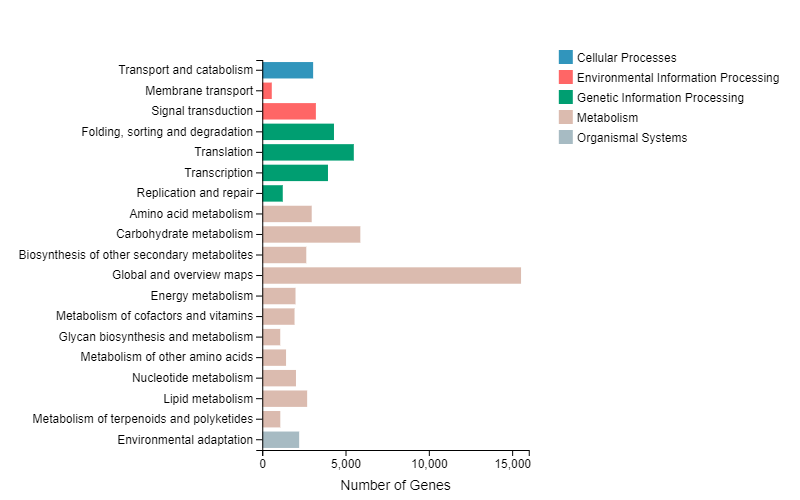
We performed GO enrichment for function classification of DEGs. The results revealed that 17 terms for biological processes, 15 terms of cellular components and 10 terms for molecular functions were concentrated in all DEGs (Supplemental Fig. S1). KEGG pathway enrichment analysis was conducted to further investigate the major regulatory pathway of DEGs. KEGG classification results indicated that DEGs were mainly enriched in cellular processes, environmental information processing, genetic information processing, metabolism and organismal systems (Supplemental Fig. S2). We focused our research on the KEGG enrichment analysis of DEGs of ST vs PEST and ST vs InTE. The DEGs for ST vs PEST mainly enriched in plant hormone signal transduction (804), phenylpropanoid biosynthesis (808), biosynthesis of amino acids (697) and glycolysis/ gluconeogenesis (382) (Fig. 2e). The DEGs for ST vs InTE mainly enriched in hormone signal transduction (802), phenylpropanoid biosynthesis (772), biosynthesis of amino acids (654) and MAPK signaling pathway (569) (Fig. 2f).
Screening of phytohormone-related DEGs regulating floral organ development
-
It has been shown that flower organ development is regulated by phytohormones such as auxin (AUX), abscisic acid (ABA), cytokinin (CK), gibberellin (GA), ethylene (ETH) and jasmonic acid (JA)[20]. In order to investigate the role of phytohormone-related genes in the regulation of stamen petaloid during double-flower development, we screened 65 DEGs involved in plant hormone biosynthesis and signal transduction pathway from all pairwise comparisons (Fig. 3). There were 34 genes involved in auxin pathways, including auxin influx carrier protein (AUX1), the auxin/indole-3-acetic acid protein (AUX/IAA), auxin-responsive protein (GH3), small auxin-up RNA (SAUR) and auxin response factor (ARF). Among them, most of the major homologs of AUX1, GH3, SAUR, ARF were downregulated in both developmental stages of stamen compared with petaloid stamen or tepal, except for Unigene22450_All (AUX1), Unigene11089_All, Unigene29394_All (SAUR), and CL6633.Contig12_All (ARF) which were upregulated. Notably, all IAA were upregulated in stamen, petaloid stamen and inner tepal phenotype, respectively. In the cytokinin signaling pathway, histidine phosphotransfer protein (AHP) and histidine kinase (AHK) were downregulated in stamen compared to petaloid stamen or tepal. A total of three genes involved in the ethylene signaling transduction pathway, among which ethylene-insensitive3 (EIN3, CL140.Contig10_All) and ethylene response factor (ERF, CL15675.Contig2_All, Unigene30251_All) were downregulated in both developmental stages of stamen compared with petaloid stamen or tepal, while ethylene receptor (ETR, CL6025.Contig6_All) and CL15628.Contig1_All (EIN3) were upregulated. Among the DEGs involved in the JA signaling pathway, the jasmonate ZIM domain-containing protein (JAZ) was expressed at a high level in both stages of stamen, whereas lipoxygenase (LOX) and MYC2 were highly expressed in petaloid stamen and tepal in contrast to the expression pattern of JAZ. In the gibberellin biosynthesis and signal pathway, the gibberellin synthesis-related gene (GA203ox, GA20ox) and GA insensitive dwarf1/2 (GID1/2) were downregulated in both stages of stamen compared with petaloid stamen or tepal. Nonetheless, the expression levels of CL452.Contig3_All (GA20ox) and Unigene10534_All (GA2ox) were upregulated. In particular, four pyrabactin resistance-like genes (PYL) involved in ABA signaling pathway were significantly downregulated in both stages of stamen compared with petaloid stamen or tepal.

Figure 3.
Expression heat maps of DEGs involved in the biosynthesis and signaling pathways phytohormone as auxin, CK: cytokinin, ETH: ethylene, JA: jasmonic acid, GA: gibberellic acid and ABA: abscisic acid. Red and blue represent up- and downregulation in gene expression, respectively. Gene expression level (log10(FPKM+1)) are represented by color gradation.
Identification of transcription factors related to stamen petaloid
-
A large number of transcription factors (TFs) were identified as DEGs in the regulation of stamen petaloid during both development stages (S1, S2). In our study, we screened out 118 TFs with |log2FC| > 2 to be significant, including MYB family with 38 members, NAC family (20), MADS family (14), Tify family (16), bHLH family (16), mTERF family (12), and bZIP family (2). In the pairwise comparison of stamen, petaloid stamen and tepal at two stages, there were 23 significant TFs amongst 118 DEGs. Their expression profiles revealed TFs that play key roles in stamen petaloid during different stages of flower development (Fig. 4a).
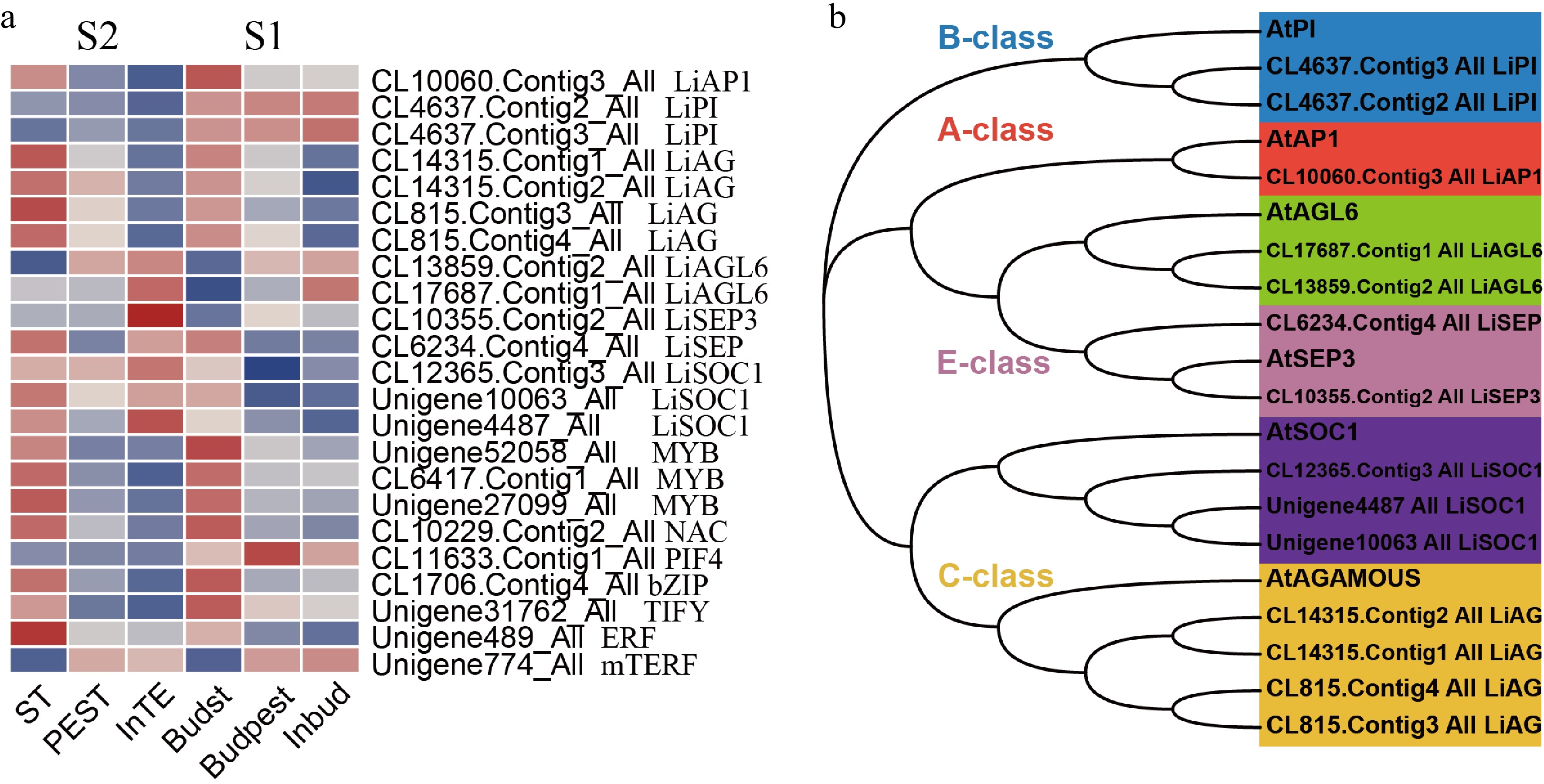
Figure 4.
Analysis of key transcription factor genes encode DEGs regulating stamen petaloid. (a) The expression heat map of key TFs in two development stages (S1, S2). (b) Phylogenetic analysis of key MADS-box family transcription factors and Arabidopsis-related proteins.
The DEGs encoding Unigene52058_All, CL6417.Contig1_All, Unigene27099_All (MYB), CL10229.Contig2_All (NAC), CL1706.Contig4_All (bZIP), Unigene31762_All (TIFY) and Unigene489_All (ERF) were downregulated in both development stages of stamen compared with petaloid stamen or tepal. In mTERF family, Unigene774_All (mTERF) was highly expressed in petaloid stamen or tepal at S1/S2 stages. bHLH family CL11633.Contig1_All (PIF4) showed higher expression levels at S1(bud stage) than S2 (full-bloom stage) and was upregulated in stamen compared with petaloid stamen or tepal. As we know, different tetramers of MIKC-type MADS-domain TFs control the identity of floral petaloid organs[10]. Therefore, we conducted phylogenetic and expression pattern analysis of key MADS-box TFs related to stamen petaloid during the development of double-flower (Fig. 4). The phylogenetic analysis showed that MADs-box TFs of Asiatic hybrid lily 'Annemarie's Dream' are classified into six subgroups as A class (SQUA/AP1), B class (AP3/PI), C class (AG) and E class (SEP/AGL2) (Fig. 4b). In the phylogenetic tree, LiAGL6 and LiSEP are clustered into one upper major clade, indicating their close evolutionary relationship, and presumably they both perform the function of E class SEP. Among 14 MADS-box TFs, LiAP1 (CL10060.Contig3_All) and LiAG (CL14315.Contig1_All, CL14315.Contig2_All, CL815.Contig3_All, CL815.Contig4_All) were significantly downregulated in both development stages of stamen compared with petaloid stamen or tepal. Meanwhile, LiAGL6 (CL13859.Contig2_All, CL17687.Contig1_All) and LiSEP3 (CL10355.Contig2_All) have similar expression patterns being upregulated in both development stages of stamen compared with petaloid stamen or tepal. In addition, LiPI (CL4637.Contig2_All, CL4637.Contig3_All) showed higher expression levels at S1 (bud stage) than S2 (full-bloom stage).
Screening for key DEGs regulating stamen petaloid through WGCNA
-
To illustrate further insight into the regulation of stamen petaloid throughout double-flower development stages, we performed WGCNA to gain the gene co-expression networks of key DEGs. A total of 29 gene co-expression modules were identified based on their similar expression profiles (Fig. 5a). Notably, we found that key DEGs in green modules displaying a distinctly different expression pattern which highly expressed in stamen in both stages of stamen but barely expressed in petaloid stamen and tepal. This may indicate the DEGs in this module are closely related to the regulation of stamen petaloid. Therefore, we identified the potential regulatory network for stamen petaloid in the green module (Fig. 5b). There were many genes related to plant hormones and TFs in this module. Firstly, two MADS-box family genes CL14315.Contig2_All (AG) and CL10060.Contig3_All (AP1) were identified as hubs within the potential regulatory network in the green module. Then, we explored the DEGs involved in TFs regulation and phytohormone signaling co-expressed with the hub genes. In this regulatory sub-network, the MYB family has the most members (6), followed by Tify (5), bZIP (4), bHLH (4), AP2-ERF (2) and NAC (1), they may both have an interaction with AG and AP1. Furthermore, we found that CL3014.Contig2_All (PYL) and CL5627.Contig1_All (GID2) as central components of ABA and GA signaling transduction respectively, that had a cross regulation with MADs-box TFs. Moreover, CL6583.Contig1_All (SAUR) and CL3630.Contig2_All (AHK) were also cross regulated by key co-expression TFs. All co-expressed genes (TFs and phytohormone-related DEGs) with MADs were downregulated in both development stages of stamen compared with petaloid stamen or tepal, which suggests that the downregulation of the key genes has a vital role in stamen petaloid during double-flower development.
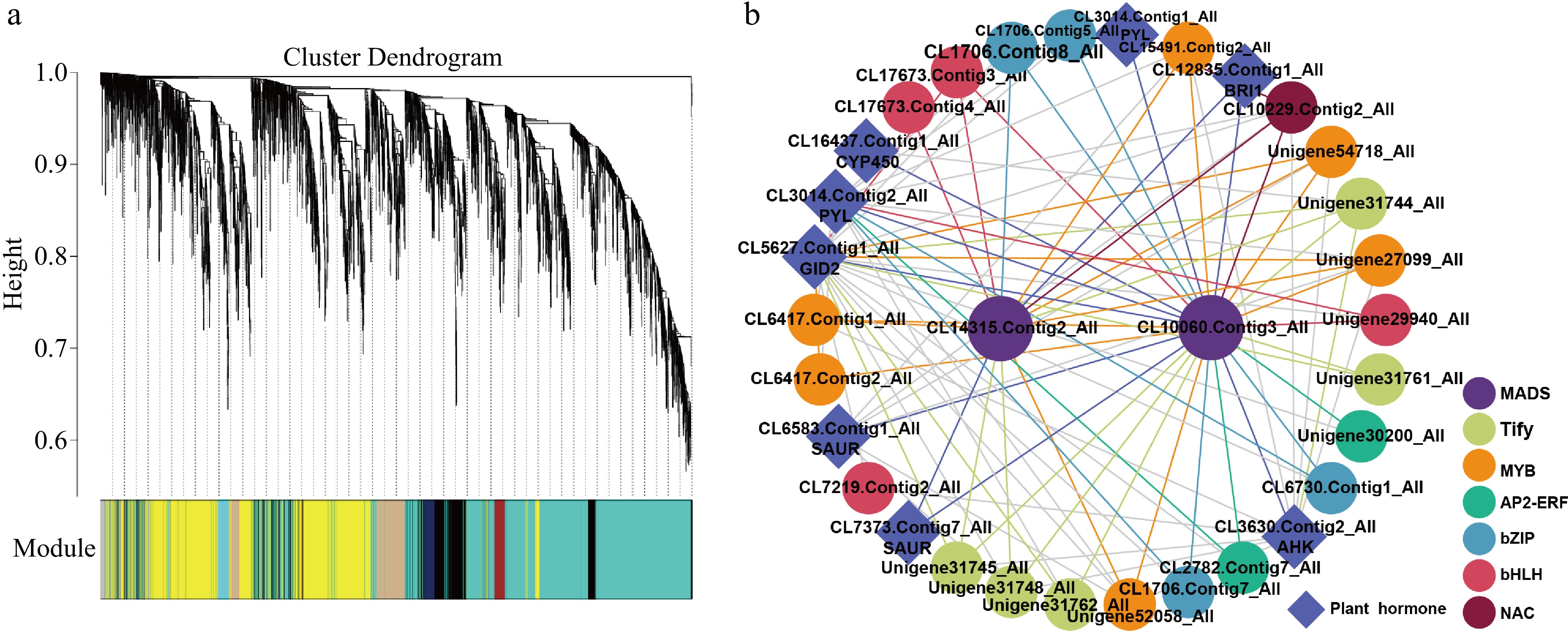
Figure 5.
The resolved gene regulatory network of stamen petalod in Asiatic hybrid 'Annemarie's Dream'. (a) Dendrogram showing co-expression modules identified by WGCNA across double-flower development. (b) The regulatory network of key genes involved in stamen petaloid.
Validation of MADs-box gene expression by qRT-PCR
-
To further verify the quality of RNA-seq data, we performed quantitative real-time PCR to validate the expression patterns of MADS-box genes. As shown in Fig. 6, the expression levels of A-class AP1, B-class PI, C-class AG, E-class SEP and AGL6 are consistent with FPKM values obtained by transcription profiles. The results demonstrate the reliability of RNA-seq data and weight gene co-expression analysis.
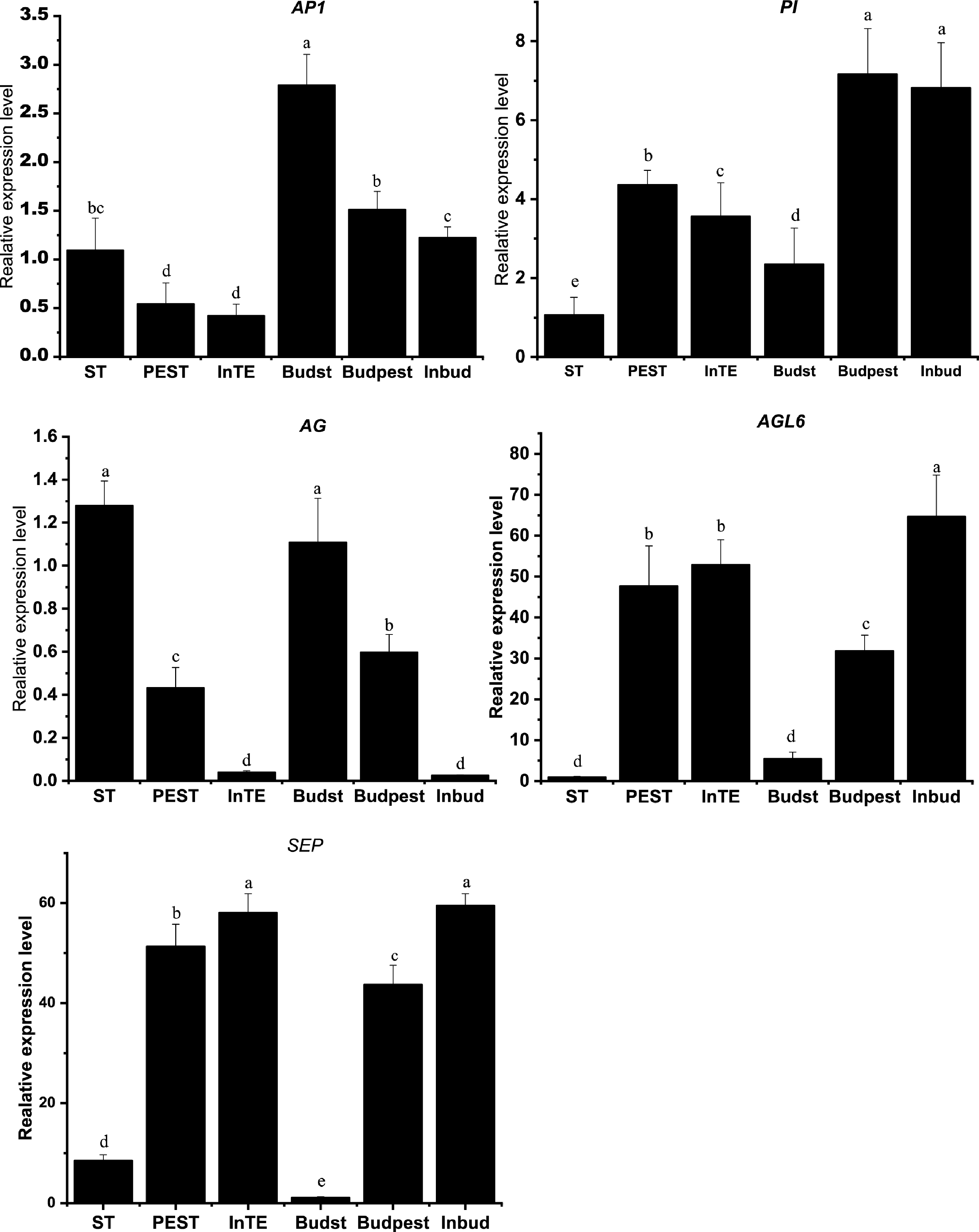
Figure 6.
Validation of the expression patterns of five MADS-box genes. Error bars represent standard deviation (SD) of biological replicates. Bars with different letters indicate significant differences among treatments, P ≤ 0.001, following one-way ANOVA.
-
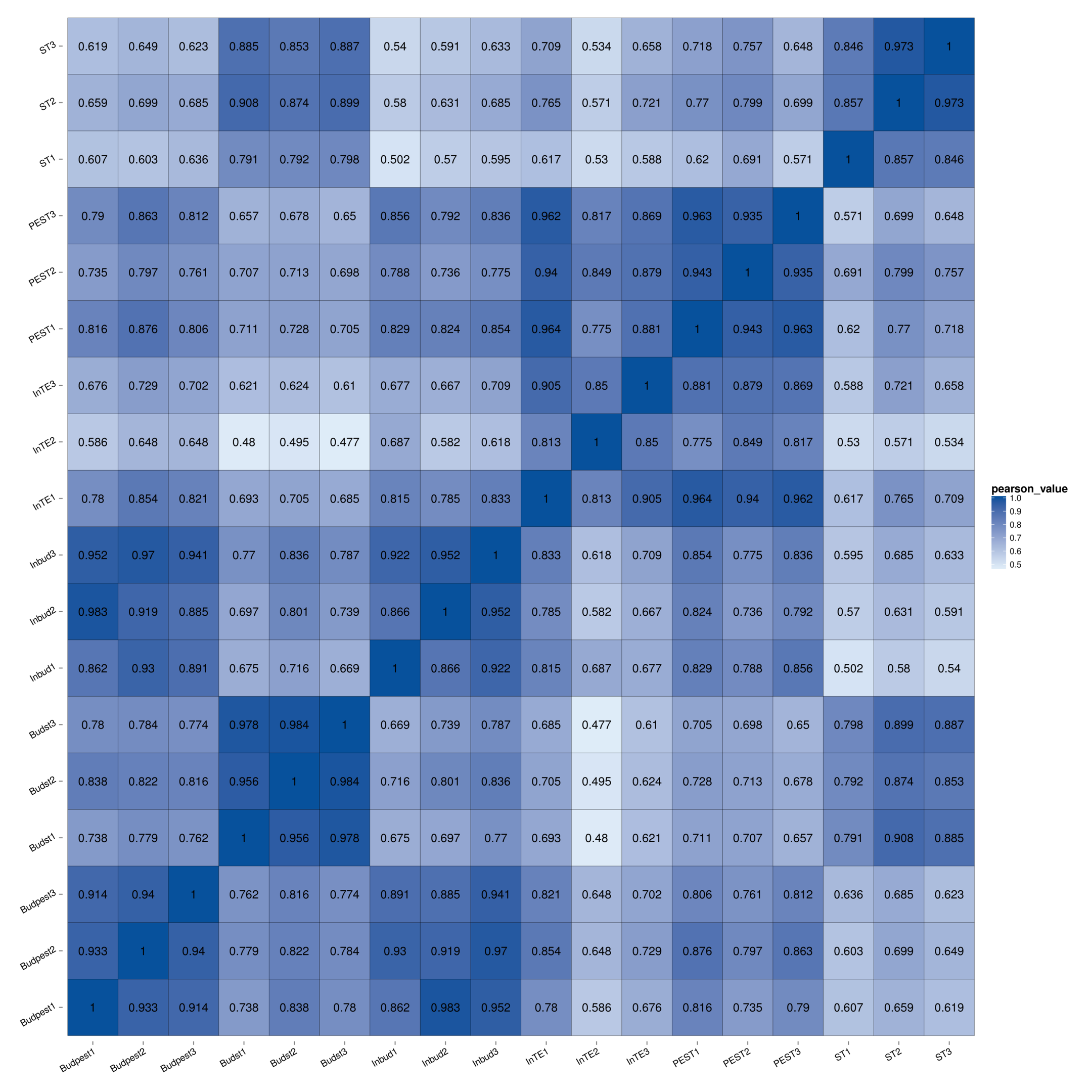
Flower type is one of the most valuable ornamental phenotypes in flowering plants. In Lilium, double-flower cultivars with petaloid stamens are of high commercial value. Based on the morphology of tepal, petaloid stamen and stamen of the Asiatic hybrid cultivar 'Annemarie's Dream', we suggest that the formation of petaloid stamen is caused by transition from stamen to tepal. To investigate the molecular mechanism underlying stamen petaloid, the comparing transcriptome analysis was conducted among stamen, petaloid stamen and tepal at two double-flower development stages. We detected 37,549 DEGs, more of which were downregulated in the comparison of stamen vs petaloid stamen and stamen vs tepal, suggesting that the down-regulation genes were significant in the transformation of stamens into petals (Fig. 2a). Moreover, we found that the number of DEGs in ST vs PEST and ST vs InTE was much larger than those in PEST vs InTE, suggesting that stamen petaloid is more like tepal than stamen. This result was further supported by the Pearson correlation coefficient analysis (Supplemental Fig. S3). Notably, the significant DEGs mainly enriched in plant hormone signal transduction (Fig. 2e), which indicates that the stamen petaloid may be regulated by a complex multilevel regulatory system. Therefore, we explored the more specific transcriptional regulatory networks containing hormone signaling gene and TFs with the help of WGCNA.
Phytohormones are involved in the regulation of stamen petaloid
-
Plant hormones are essential factors affecting flower organ development[23]. In our study, there were numerous phytohormone-related genes involved in biosynthesis and signaling of auxin, cytokinin, ethylene, jasmonic acid, gibberellic acid and abscisic acid, which were differentially expressed across stamen, petaloid stamen and tepal. Among them, biosynthesis and signaling genes of auxin and cytokinin are involved in the formation of floral organs and cell growth and proliferation of petals during flower development[24−26]. In our study, the signaling related genes of auxin (AUX1, GH3, SAUR and ARF) and cytokinin (AHK, AHP) were almost downregulated in both stages of stamen compared with petaloid stamen or tepal, indicating its important role in determining the flora organ identity in Asiatic hybrids (Fig. 7). Previous studies have confirmed that auxin has significant activity during plant reproductive development, especially in anthers and filaments[27,28]. Therefore, we inferred auxin may be synthesized more in stamen than in stamen petaloid/tepal. However, we found all IAA genes showed higher expression level in petaloid stamen and tepal than stamen, indicated that these genes as auxin downstream response factors might play important roles in stamen petaloid. It has been shown that when the IAA8 protein is mutated in Arabidopsis, the transgenic plants have abnormally floral organs with short petals and stamens[29]. Furthermore, IAA8 plays its role in the development of floral organs by changes in JA levels probably via its interaction with ARF6/8 proteins, since both ARF6/8 are required for normal JA (jasmonic acid) production. Other studies have also confirmed that hydrogen peroxide dehydrogenase (AOS) and hydrolase synthesis (HPL) together facilitate the metabolism of JA and may lead to the maturation of petaloid stamens[21]. Thus, we hypothesized that that IAA and JA play an important role in regulating flora organ development and promoting the formation of petaloid stamens. Our transcriptome datasets confirmed that most of the genes related to the JA signaling pathway showed a higher expression level in petaloid stamen and tepal than stamen. In addition, PYL genes as ABA signaling receptor were downregulated in both stages of stamen compared with tepal and petaloid stamen, indicated that stamen petaloid is controlled by a complex network of hormonal regulation.

Figure 7.
Summary of candidate DEGs involved in phytohormone signals and biosynthesis regulating stamen petaloid in the Lilium cultivar ‘Annemarie’s Dream’. The vertical up arrow represents the upregulated genes from stamen to petaloid stamen phenotype. The vertical down arrow represents the downregulated genes from stamen to petaloid stamen.
Involvement of transcription factors in the formation of petaloid stamen
-
Many TF family members, such as MYB, NAC, bZIP, TIFY, bHLH, ERF and mTERF showed significantly distinct expression patterns among stamen, petaloid stamen and tepal at two development stages, suggesting they may participate in the regulation of stamen petaloid. Previous studies have confirmed that GA can promote JA production and high levels of JA can induce MYB expression and thus promote stamen development. Furthermore, MYB has been reported to play a critical role in the development of floral organs and pollens[30−32]. In this study, we found that most of the transcription factors were highly expressed in both stages of the stamen, with the largest number of MYB families, so we speculated that key MYB genes might be involved in stamen formation. In addition, the NAC transcription factor gene can be rapidly induced by ethylene and is involved in the regulation of petal size and floral boundary by activating or inhibiting the cell expansion of petals[33,34]. CL10229.Contig2_All, which was identified as NAC-like TFs, is likely involved in stamen development. It has been suggested that mTERF, Tify, MYB-related, bHLH and NAC TFs are probably involved in flower primordium differentiation such as perianth differentiation and stamen differentiation in Erythronium japonicum[35]. bHLH TFs may mediate region, organ, and floral type specific signals in L. speciosa inflorescences[36]. Meanwhile, the abundance of bHLH family TFs PIF4 was regulated by BLADE-ON-PETIOLE (BOP) genes, which have previously been proven to control leaf and flower development in Arabidopsis[37]. We found that CL11633.Contig1_All (PIF4) was upregulated in stamen compared with petaloid stamen or tepal and showed higher expression levels at S1(bud stage) than S2 (full-bloom stage), which suggests that it may play a role in promoting the flowering process and floral organ identity.
MADs-box DEGs involved in stamen petaloid
-
The MADS-box transcription factor plays an important role in plant reproductive development, especially as its homologous proteins are in a pivotal position in the floral organ identity. According to the 'ABCE' model, four whorls of floral organs are regulated by tetrameric protein complexes of MIKC-type MADS-domain transcription factors A-, B-, C- and E-class[10]. Therefore, we focus on the expression patterns of MADS-box genes in different floral tissues during double-flower development. An increasing number of studies have shown that altered expression patterns of AG homologs lead to stamen petaloid in double-flower phenotypes of non-model plants. In our study, we screened 14 significantly DEGs from MADS-box TFs and classified them into four A-, B-, C-, and E-class based on their phylogenetic analysis with Arabidopsis-related genes. Among them, four homeotic LiAG genes showed highest expression levels in stamen compared to petaloid stamen and tepal. Similarly, the decreased expression of the AG ortholog PrseAG led to double-flower formation in Prunus lannesiana[38]. Ectopic expression of EjAG identified in double-flower loquat (Eriobotrya japonica) rescued the development of stamens and carpels from the double-flower phenotype in an Arabidopsis ag mutant[23]. In summary, these studies suggest the destruction of floral organ-determining gene AG may lead to the homeotic transformation of stamen into tepal. In addition, we found that LiAGL6 and E-class gene LiSEP3 were upregulated in both development stages of stamen vs petaloid stamen or tepal, in contrast to the expression pattern of LiAG. That indicates that LiAGL6 and LiSEP3 are involved in the transformation of stamen to tepal.
Mao et al.[39] applied a strategy using in vivo fluorescence resonance energy transfer (FRET) to find complicated tepal and stamen heterotetrameric complexes in lily and verified lily (Lilium longiflorum). PI co-orthologs LMADS8 and LMADS9 likely formed heterotetrameric petal complexes with Arabidopsis AP3/SEP3/AP1, which rescued petal defects of pi mutants. However, LMADS8 and LMADS9 did not form heterotetrameric stamen complexes with Arabidopsis AP3/SEP3/AG to rescue the stamen defects of the pi mutants. In our study, B-class gene LiPI were highly expressed in bud stages and showed higher expression levels in stamen petaloid and tepal than stamen, which indicated that LiPI may form a tetramer with AP1, AP3 and SEP3 to regulate the formation of petaloid stamen.
Through WGCNA analysis, we discovered the green gene co-expression module that may be involved in the regulation of stamen petaloid. Two MADS-box TFs (LiAG, CL14315.Contig2_All; LiAP1, CL10060.Contig3_All) were identified as hub genes in this module. Centering on these genes, we identified key co-expressed genes that determined floral organ development at a transcriptional level, specifically the stamen petaloid. These genes form a multi-level regulatory network involving auxin, GA and ABA signaling and some key TFs such as MYB. Our transcriptome data provides an insight into the molecular regulatory network underlying stamen petaloid and thereby offering a theoretical basis for double-flower breeding in Lilium.
-
Asiatic Hybrids lily 'Annemarie's Dream' cultivars were grown in the nursery of Badaling Forest Farm (Beijing, China). The double-flower cultivars were cultivated in the greenhouse and three parts of the floral organ were collected from the bud stage to the full-bloom stage. Bud stage: Inner bud (Inbud), petaloid stamen (Budpest) and staminode (Budst). Full-bloom stage: Inner tepal (InTE), petaloid stamen (PEST) and staminode (ST). Each sample was obtained from nine flowers or flower buds at two development stages, three of which were used as one biological replicate, for a total of three biological replicates. Samples were immediately flash frozen in liquid nitrogen and stored at −80 °C for RNA extraction.
RNA extraction, cDNA library construction and Illumina sequencing
-
Extraction of total RNA from six samples, including three biological replicates were carried out using RNAprep pure Plant Kit (TIANGEN Biotech, Beijing, China) and the RNA integrity number (RIN) of each sample needed to be > 7.3 for cDNA library construction. mRNA with polyA tail was enriched by Oligo (dT) beads and rRNA was removed using DNA hybridization probes. Subsequently the broken short mRNA fragment was used as a template to create cDNA libraries and library quality was assessed on the Agilent 2100 Bioanalyzer and ABI Step One Plus Real-Time PCR System. Illumina sequencing was performed at Illumina Hiseq platform by BGI Co. (Beijing, China).
Transcriptome data processing and gene functional annotation
-
Clean reads were obtained by removing low-quality reads, reads containing adapters and poly N and reads with unknown base 'N' content greater than 5%. De novo assembly of clean reads using Trinity v2.0.6 was performed[40], followed by TGICL[41] to cluster the assembled transcripts and remove redundancy to obtain Unigene. Transdecoder v3.0.1 was used to identify candidate coding regions in Unigene by aligning the homologous protein sequences in the SwissProt or Pfam database[42]. For gene functional annotation, the assembled unigene were aligned and annotated using HMMER v3.0[43], BLAST v2.2.23[44], and BLAST 2GO v2.5.0 to seven functional databases as GO (Gene Ontology), KEGG (Kyoto Encyclopedia of Genes and Genomes), NR (Non-redundant proteins), NT (Nucleotide sequence database), COG (clusters of orthologous groups), Swiss-Prot, and Pfam.
Analysis of differential expression genes
-
The gene expression level of each sample was calculated by RSEM v1.2.8[45], based on Fragments Per Kilobase of transcript per Million (FPKM). Differential expression gene analysis was performed based on Poisson distribution, DEGs detection was performed according to the method described in Wang et al.[46]. In order to improve the significance of DEGs, P-values are corrected to Q-values using the strategy employed by Storey & Tibshirani[47]. In this study, we defined genes at Fold Change > 2, Q-value ≤ 0.001 and false discovery rate (FDR) < 0.05 in a comparison were recognized as significant differentially expressed genes.
Quantitative real-time PCR validation of RNA-seq data
-
Validation of RNA-seq data related to MADS-box TFs using qRT-PCR. The qRT-PCR reactions were performed on the iQTM5 using SYBR. The primers used in this study are listed in Supplemental Table S2. The PCR protocol was initiated at 94 °C for 3 min, followed by 40 cycles of 94 °C for 20 s, 60 °C for 30 s and 72 °C for 30 s. CT values (Cycle threshold) were recorded after completing 40 cycles. The data was obtained from three biological replicates, each of which contains three technical replicates. Relative gene expression was normalized with the lily actin gene as an internal reference and was analyzed using the 2−ΔΔCᴛ method[48]. The difference of the mean values for the different treatments were compared by post-hoc least significant difference tests. Values of P < 0.001 were considered to indicate significance. Origin software is used for chart drawing in Fig. 6.
Weighted correlation network analysis
-
After discarding relative low expression genes (the FPKM was less than 1 in more than 18 samples), the R package WGCNA[49] was used to identify modules of highly co-correlated gene modules base on the filtered FPKM data. The co-expression modules were obtained using automatic network construction function (block wise modules) with power = 15, minModuleSize = 100, TOMtype was signed. Eigengene value was calculated for each module based on Pearson correlation. The networks were visualized by Cytoscape (v.3.8.2)[50].
This work was supported by grants from the Opening Foundation of Beijing Engineering Research Center of Rural Landscape Planning and Design (KF2020).
-
The authors declare that they have no conflict of interest.
- Supplemental Table S1 The quality and quantity of transcriptome sequencing data.
- Supplemental Table S2 The primer sequences required for qRT-PCR.
- Supplemental Fig. S1 GO function classification map.
- Supplemental Fig. S2 KEGG function classification map.
- Supplemental Fig. S3 Pearson correlation of genes expression in staminode, petaloid stamen and inner tepal at bud and full-bloom stage.
- Copyright: © 2022 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Wang W, He X, Li X, Wang W. 2022. Transcriptome profiling during double-flower development provides insight into stamen petaloid in cultivated Lilium. Ornamental Plant Research 2:10 doi: 10.48130/OPR-2022-0010 

Transcriptome profiling during double-flower development provides insight into stamen petaloid in cultivated Lilium
- Received Date: 23 April 2022
- Accepted Date: 16 June 2022
- Published Online: 30 June 2022
Abstract: Asiatic hybrid lilies (Lilium spp.), as the biggest cultivar groups of the lily variety are an ornamental plant with elegant floral patterns and bright colors. We discovered a double-flower Asiatic hybrid cultivar ‘Annemarie’s Dream’ whose stamens convert into petaloid stamens in varying degrees. Double-flower is a significant ornamental trait of the flower organ. However, the molecular mechanism of stamen petaloid formation has not been widely studied in Asiatic hybrid lily. Therefore, we used RNA-seq to contrast transcriptomes of stamen, petaloid stamen and inner tepal at two developmental stages. In total, 190,488 unigenes were obtained and 37,549 differentially expressed genes (DEGs) were identified. We focused on DEGs involved in phytohormone signaling and transcription factor regulation, especially the MADS-box genes (A-class gene LiAP1; B-class gene LiPI; C-class gene LiAG; E-class gene LiAGL6, LiSEP3). Furthermore, we performed weight gene co-expression network analysis (WGCNA) and identified two co-expressed MADS-box homeotic genes (LiAG, CL14315.Contig2_All; LiAP1, CL10060.Contig3_All) as hubs. We also found that CL3014.Contig2_All (PYL) and CL5627.Contig1_All (GID2) as phytohormone-related genes may participate in the regulation of the stamen petaloid during double-flower development. In summary, our findings provide an insight into the molecular regulatory network underlying stamen petaloid and thereby offering a theoretical basis for double-flower breeding in Lilium.
-
Key words:
- Trancriptome /
- Asiatic hybrid lily /
- Stamen petaloid /
- WGCNA
{kind=link}
{kind=link}
{kind=link}