Evidence of Elevational Speciation in Kerteszia cruzii (Diptera: Culicidae) in the Ribeira Valley, São Paulo, Brazil
 Bruna Demari-Silva1*
Bruna Demari-Silva1*  Gabriel Zorello Laporta2
Gabriel Zorello Laporta2  Tatiane Marques Porangaba de Oliveira1
Tatiane Marques Porangaba de Oliveira1  Maria Anice Mureb Sallum1
Maria Anice Mureb Sallum1- 1Faculdade de Saúde Pública, Departamento de Epidemiologia, Universidade de São Paulo, São Paulo, Brazil
- 2Centro Universitário FMABC da Fundação ABC, Setor de Pós-graduação, Pesquisa e Inovação, Santo André, Brazil
Kerteszia cruzii [former Anopheles (Kerteszia) cruzii] is a bromeliad mosquito widespread in the Brazilian Atlantic rainforest. In South-eastern Brazil, it plays an important role in malaria transmission because it was infected with at least four Plasmodium species. There is robust evidence that Ke. cruzii is a species complex. We used single nucleotide polymorphisms (SNPs) from a nextRAD sequence (nextera-tagmented, reductively amplified DNA) to investigate the genetic structure of Ke. cruzii in the Ribeira Valley, South-eastern Brazil. Furthermore, we verified whether the genetic structure was associated with forest cover, elevation, slope, and vegetation physiognomy. Our results showed two distinct lineages in the studied region associated with elevation and isolation by distance. The first lineage included samples from coastal localities and the second comprised specimens from inland or mountain sites. At one sampling locality (Esteiro do Morro in Cananéia municipality), both lineages are sympatric. These results are in accordance with previously published data that showed elevated stratification in Ke. cruzii. However, Fst values did not indicate the existence of cryptic or sister species in Ke. cruzii in this region, we concluded that elevational speciation probably occurs, and we hypothesized that differences in population structure found might be associated with the distribution of bromeliad species.
Introduction
Kerteszia cruzii occurs in areas of the Brazilian Atlantic rainforest and is abundant, depending on the abundance of the bromeliad phytotelmata. Formerly, Kerteszia was classified as a subgenus of the genus Anopheles until 2017, when the results of a phylogenetic analysis of the complete mitochondrial genome consistently showed Kerteszia as a monophyletic taxon placed outside the genus Anopheles (Foster et al., 2017). Consequently, Foster et al. (2017) elevated Kerteszia, Lophopodomyia, Stethomyia, and Nyssorhynchus to the genus level.
Females of Ke. cruzii are primarily sylvatic, presenting a low degree of sinanthropy (Forattini, 2002), can feed on humans, and are more abundant at the edges of forests where they usually perform their blood repast (Forattini et al., 1986; Medeiros-Sousa et al., 2019). Although the females of some populations are capable of blood-feeding at any time of the day, they usually present two activity peaks, one at twilight and the other at dawn (Forattini et al., 1986). Females that feed at dusk were found to live longer and, therefore, are more likely to survive and exceed the extrinsic incubation period of Plasmodium (Dalla Bona and Navarro-Silva, 2010).
The epidemiological importance of this species in the Atlantic Forest is unquestionable. In the seventies, this species was naturally infected with Plasmodium brasilianum and Plasmodium simium (Deane et al., 1970). Ever since, Ke. cruzii was found naturally infected with Plasmodium vivax and P. vivax VK247, Plasmodium falciparum, and Plasmodium malariae in São Paulo and Espírito Santo states, Brazil (Branquinho et al., 1997; Duarte et al., 2013; Kirchgatter et al., 2014; Laporta et al., 2015; Buery et al., 2018; Demari-Silva et al., 2020). Recently, studies have demonstrated that P. vivax and P. simium are almost indistinguishable and can infect humans (Brasil et al., 2017; de Alvarenga et al., 2018).
Several studies have suggested that Ke. cruzii is a complex of species. Analyses of the banding pattern of the ovarian polytene chromosome showed that this species encompasses three sibling species in the Boracéia and Juquitiba municipalities in São Paulo state, Brazil (Ramírez and Dessen, 2000a; Ramírez and Dessen, 2000b). A broader study using samples from Rio de Janeiro (RJ), São Paulo (SP), Santa Catarina (SC), and Bahia (BA) states in Brazil demonstrated that the specimens from Bahia state are genetically distant from the remaining populations analyzed from the South and Southeast of Brazil (Carvalho-Pinto and Lourenço-de-Oliveira, 2004).
Subsequently, studies employing the timeless nuclear gene reaffirmed the findings of Carvalho-Pinto and Lourenço-de-Oliveira (2004) and suggested that in Itatiaia municipality, RJ state, Brazil, there are two putative species under the name of Ke. cruzii (Rona et al., 2010, 2013). A study in Cananéia municipality in Southeastern Brazil employing wing geometric morphometrics and COI gene analyses showed that samples from hilltops differ from those found in the lowlands (Lorenz et al., 2014). Analyses employing the complete mitochondrial genome of four Kerteszia specimens, including four distinct populations of K. cruzii, showed differences in the codon composition from three localities in the South-eastern region (São Paulo and Cananéia both in São Paulo state; Itatiaia in RJ) of Brazil and one from South Brazil (Maquiné in the Rio Grande do Sul state) (Oliveira et al., 2016). Analyses using the cpr and clock nuclear genes revealed two lineages in the Serra do Mar region: one corresponding to samples from the low land coast and another corresponding to that from mountain specimens (de Rezende Dias et al., 2018). Recently, Kirchgatter et al. (2020), employing sequences from GenBank from NADH4 and COI, identified three genetic lineages of this species in Brazil corresponding to the Serra do Mar, Serra da Mantiqueira, and Serra da Cantareira.
Herein, we employed single nucleotide polymorphisms (SNPs) from a nextRAD sequencing (nextera-tagmented, reductively amplified DNA) to verify whether landscapes presenting heterogeneous vegetation physiognomies, with distinct vegetal coverage, can influence the genetic structure of Ke. cruzii. The main objectives of this study were to (1) test the genetic structure of Ke. cruzii in different gradients of vegetation cover in six locations described in Laporta et al. (2015); (2) to search for genetic signatures associated with distinct environmental variables.
Materials and Methods
Mosquito Collection and DNA Extraction
Mosquitoes were collected between July 2016 and December 2018 (Table 1). Ke. cruzii specimens were collected from seven field collection sites as follows: (1) Tapiraí, (2) Sete Barras, (3) Eldorado, (4) Eldorado – Toca da Onça, (5) Cananéia, Esteiro do Morro, (6) Cananéia, Sítio Itapuan, and (7) Ilha Comprida, Pedrinhas. The longitude and latitude (GPS, datum WGS84) data were obtained from each collection site and georeferenced in the Geographic Information System (ArcGIS v. 10.3.1, and QGIS v. 2.18.9) in EPSG: 4326 (WGS84, world geographic projection). The elevation map was obtained from the Shuttle Radar Topography Mission (SRTM) 1.4, interpolated to a 30-m spatial resolution. An elevation map was used to construct the terrain slope topographic layers of the study region. These topographic layers were overlapped and projected in GIS together with vegetation cover layers (i.e., ombrophilous dense forest, restinga, and mangrove) obtained from SOS Mata Atlântica/INPE and Landsat images (30-m spatial resolution).

Table 1. Field collection information.
The collection sites in Esteiro do Morro, Eldorado, Sete Barras, Toca da Onça, and Tapiraí are in transition landscapes between dense ombrophilous forests and rural environments, with forest cover gradients of 74.99% (Tapiraí), 65.37% (Esteiro do Morro and Sete Barras), and 44.66% (Eldorado). Sítio Itapuan and Pedrinhas presented heterogeneous landscapes. For example, in Sítio Itapuan, besides dense ombrophilous forest and the rural environment, restinga, water, and mangroves (97.49% of natural Atlantic Forest) are still present. The landscape in Pedrinhas is characterized as a transition between the urban environment, restinga vegetation, and mangroves (with 94.48% of the preserved natural vegetation) (Laporta et al., 2015) (Figure 1).
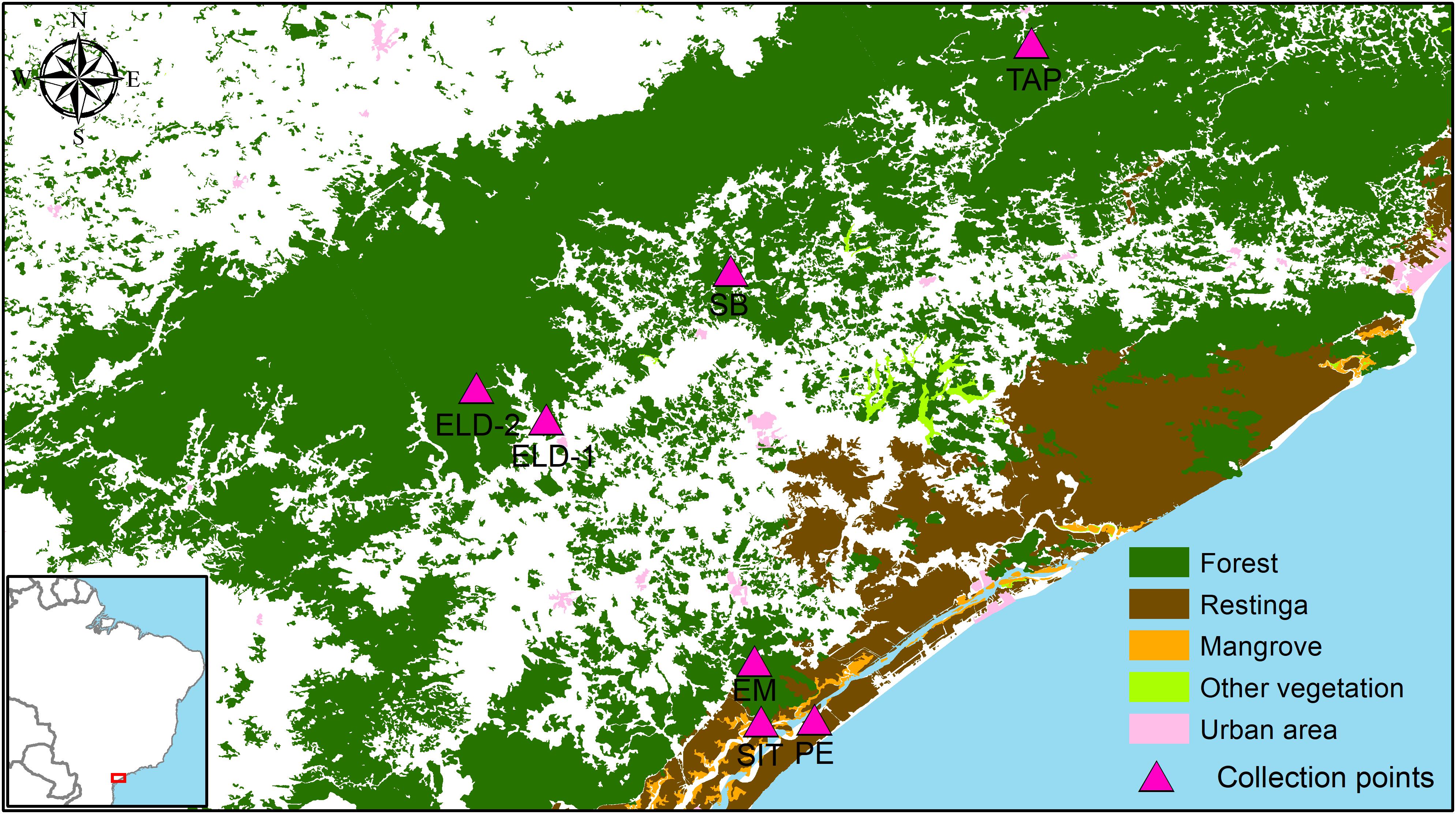
Figure 1. Study region: Ribeira Valley, Southeastern São Paulo State, Brazil. Field collection points were utilized for sampling from mosquito fauna in remnants of the Brazilian Atlantic rainforest conserved in the study region. Source: SOS Mata Atlântica/INPE.
Field collections were performed using Shannon traps from 6:00 p.m. to 10:00 p.m. The mosquitoes were preserved in silica gel until morphological identification and DNA extraction. Specimens were morphologically identified using the dichotomous keys of Forattini (2002) and Consoli and Lourenço-de-Oliveira (1994) and confirmed by barcode COI amplification and sequencing (Bourke et al., 2018). Genomic DNA was extracted using the salt method described by Miller et al. (2007) and modified by Laporta et al. (2015) from 59 mosquito specimens (Table 1).
Next Generation Sequencing (NGS) and SNPs Detection
The nextRAD libraries were assessed by the SNPsaurus LLC Company, as in Russello et al. (2015). Briefly, the genomic DNA was first fragmented using Nextera reagent (Illumina, Inc.), which also ligated short adapters to the ends of the fragments; the reaction was scaled to fragment 3 ng of genomic DNA. Each fragment was then amplified using 25 cycles at 75°C, with one of the primers matching the adapter and extending eight nucleotides into the genomic DNA with the selective sequence TGCAGGAG. Thus, only fragments starting with a sequence that can be hybridized by the selective sequence of the primer can be efficiently amplified. Thereafter, the amplified fragments were sequenced on a HiSeq 4000 with one lane of 150 bp reads (University of Oregon).
Genotyping analysis was performed with custom scripts (SNPsaurus, LLC) using bbduk (BBMap tools)1 to trim the reads (Supplementary Material S1). A de novo reference was created after collecting 10 million reads equally from the samples; reads with counts less than 7 or greater than 700 were excluded from the analysis. The remaining loci were aligned to each other to identify allelic loci and collapsed allelic haplotypes to a single representative. Then, the reads were mapped to match the reference with a threshold of 95% using bbmap (BBmap tools), and genotype calling was performed with call variants in BBmap tools. Finally, the vcf archive was filtered using vcf tools (Danecek et al., 2011) to exclude: (1) alleles with frequencies less than 5%; (2) genotype calls under 50%; (3) samples with more than 50% missing data; (4) loci with more than 10% missing data; and (5) loci with a deviation of the Hardy-Weinberg equilibrium (P < 0.01).
Population Analyses
The vcf file was converted into the necessary formats to perform the remaining analyses in PGDspider v 2.0 (Lischer and Excoffier, 2012). To assess the genetic distances within individuals and groups of individuals, two matrices were generated using Nei’s distances in R package v. 3.5.2, using StAMPP (Pembleton et al., 2013). The matrices were visualized in Splitstree4 v. 4.14.2 (Huson and Bryant, 2006). StAMPP was also employed to generate pairwise Weir and Cockerham’s (1984) Fst matrixes. The statistical significance (P) of each value was determined using 100 permutation tests. To evaluate the genetic structure of the studied samples, Bayesian analysis in STRUCTURE software v. 2.3.4 (Pritchard et al., 2000) and principal component analysis (PCA) in R v. 3.5.1, using the adegenet package (Jombart, 2008; Jombart and Ahmed, 2011) was performed. STRUCTURE analysis was performed using Strauto (Chhatre and Emerson, 2017) in seven runs (K = 1–7) with ten replicates for each run. The Markov chain Monte Carlo (MCMC) was carried out for 1 million generations and a burn-in period of 100,000 for each run. The Evanno method (Evanno et al., 2005) was implemented in STRUCTURE Harvester (Earl and vonHoldt, 2012) to determine a suitable number of clusters. The ten replicates for the best K value were combined in CLUMPP v1.1.2 (Jakobsson and Rosenberg, 2007) under the algorithm Large K Greedy with 2,000 permutations, and the results were visualized in Distruct v 1.1 (Rosenberg, 2004). Both CLUMPP and distruct were used through the pipeline CLUMPAK (Kopelman et al., 2015) on the webserver http://clumpak.tau.ac.il. The K-means clustering of the PCA analysis was determined for the Bayesian inference criterion (BIC), and then, we performed a discriminant PCA (dPCA) analysis.
Arlequin v. 3.5 software (Excoffier and Lischer, 2010) was employed to evaluate isolation by distance with 1,000 permutation tests and to perform a molecular variance analysis (AMOVA) (Weir and Cockerham, 1984) with 1,000 permutations. The latter was implemented in two ways: one considering the seven localities belonging to one group. At the same time, in the second, the populations were divided into two groups: one containing the lowland samples and the other containing the samples collected in the interior and mountains.
Genomic Signatures
We employed two distinct methodologies to detect the candidate loci. The first is based on differences in allelic frequencies in samples implemented by BayScan v 2.01 (Foll and Gaggiotti, 2008). This software uses the multinomial model Dirichlet and the reversible jump Markov chain Monte Carlo (RJ-MCMC) algorithm to obtain the posterior probability distributions. We used the default to perform this analysis, which uses 20 pilot runs with 5,000 interactions to adjust the distribution of the RJ-MCMC algorithm and a false discovery rate (FDR) value of 0.05.
The second approach was implemented in LFMM v.1.2 (Frichot et al., 2013). This methodology associates the allelic frequencies with environmental variables, using latent factor mixed models based on a Bayesian distribution, which can decrease FDR because it can estimate aleatory effects, which may be associated with genetic population events and isolation by distance. The number of latent factors was based on the STRUCTURE, PCA, and dPCA results. To decrease the FDR rates, we estimated the inflation factor according to the authors’ suggestions. Based on the hypotheses of the study, the following variables were quantified in the landscape (100-km2) surrounding the field collection points: (1) the mean elevation, (2) the mean terrain slope, and (3) the proportion of each vegetation cover (i.e., ombrophilous dense forest, restinga, and mangrove) (Supplementary Material S2). In addition to these variables, the distance (km) from the field collection point to the coast shore was also calculated (Table 2).
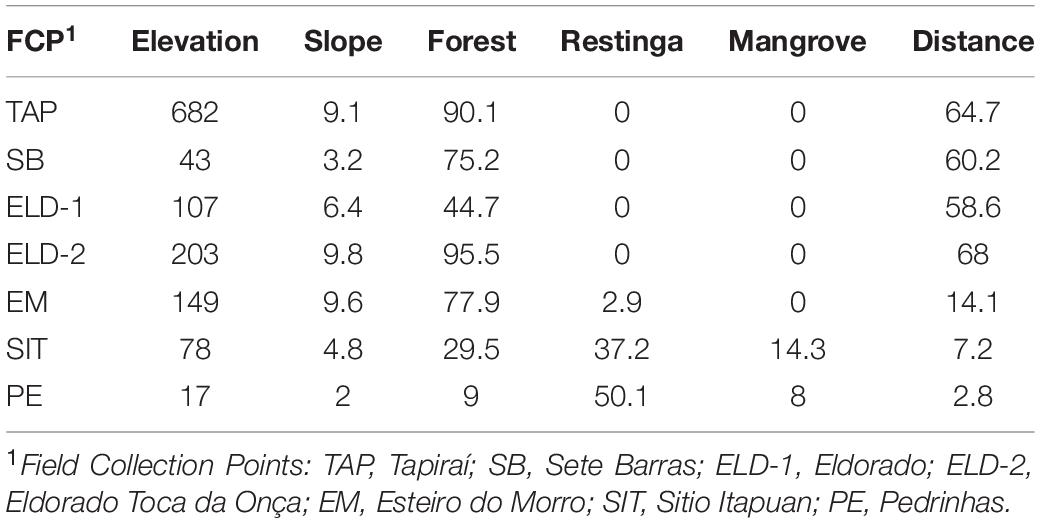
Table 2. Field collection points and environmental variables.
Results
After filtering, 4,523 loci per individual remained out of the original 19,906 loci from the genotype call. In addition, one sample from Sete Barras showed more than 50% missing data; thus, it was discarded from further analyses. The unrooted phylogenetic tree (Figure 2A), produced using the Nei’s distance matrix with the individuals, showed two distinct groups: one with only lowland samples (from Pedrinhas – Ilha Comprida; Esteiro do Morro and Sítio Itapuan, both in Cananéia municipality), and another with all individuals from the inland (Tapiraí, Sete Barras, Eldorado, and Eldorado – Toca da Onça), and two individuals from Esteiro do Morro. The second network recovered using the same analytical approach as in the first round, but defining the populations of all seven sites sampled (Figure 2B), showed three distinct genetic groups. The first group included all specimens from inland sites, the second group included the Esteiro do Morro population only, and the third group consisted of the remaining lowland samples (Pedrinhas and Sítio Itapuan).
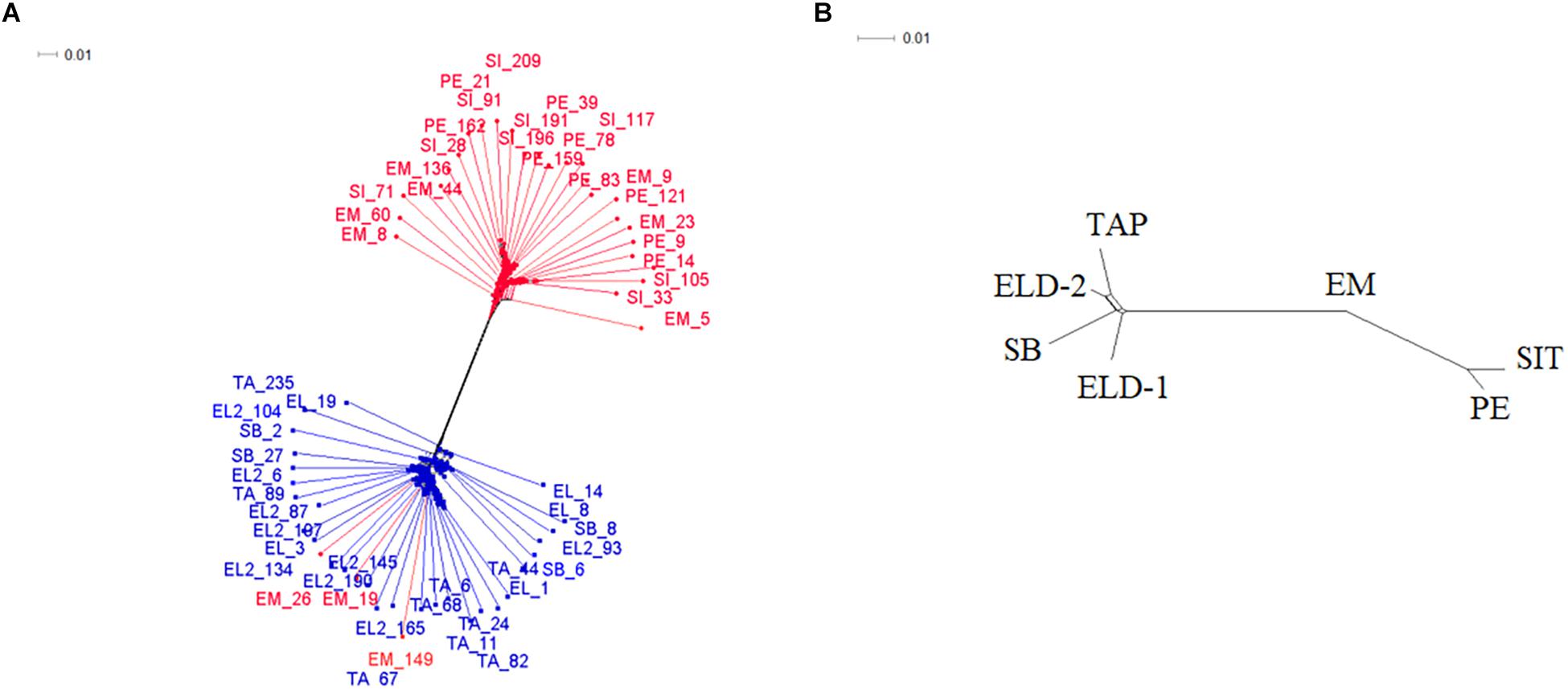
Figure 2. Decomposition network generated in SplitsTree4 software from the Nei’s distance matrix. (A) Network yielded from each individual employed in the study; the representatives in blue are those from inland/mountain regions and those in red are from lowland regions. (B) Network considering seven populations a priori. SI/SIT: Sítio Itapuan; EM: Esteiro do Morro; PE: Pedrinhas; TA/TAP: Tapiraí; EL/ELD-1: Eldorado; EL2/ELD-2: Eldorado Toca da Onça; SB: Sete Barras.
The pairwise divergence (FST) results were relatively low. However, they were statistically significant, except between Eldorado and Sete Barras (Table 3). The distances ranged from 0.001 (between Eldorado and Sete Barras populations) and 0.279 (between the Sítio Itapuan and Tapiraí populations). Among lowland specimens, the Fst values varied from 0.01 to 0.3, and the countryside samples ranged between 0.001 and 0.038. The best-fit K chosen by the Evanno method was K = 2. The Bayesian multilocus analysis from STRUCTURE showed Esteiro do Morro as the most heterogeneous population, while Sítio Itapuan was the most homogenous (Figure 3).
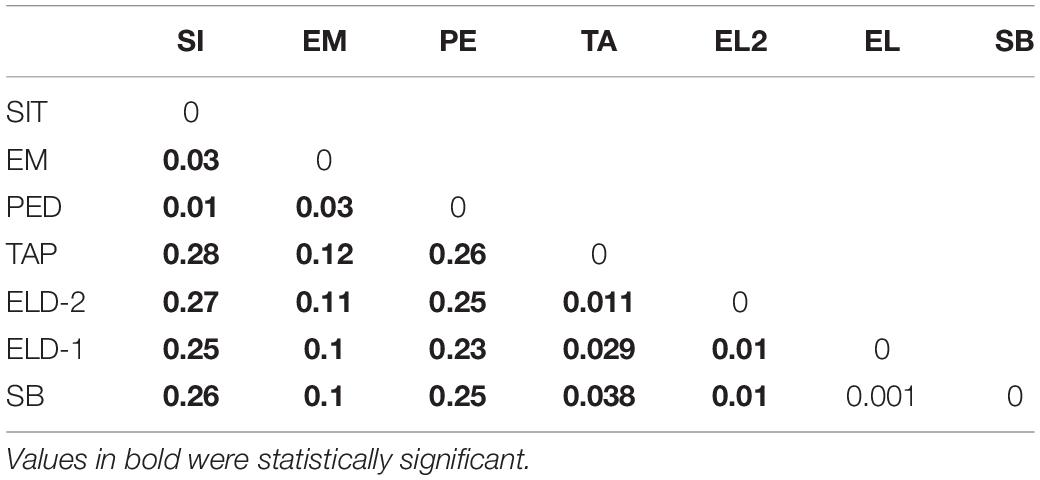
Table 3. Matrix of estimate of the genetic heterogeneity (Fst).
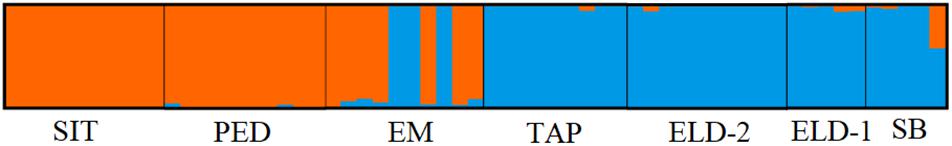
Figure 3. STRUCTURE software result showing the two genetic clusters inferred by the Evanno method.
The first two principal components represent only 25% of the variability. However, the analysis showed the same tendency of Nei’s distance and the Bayesian analysis of STRUCTURE software. The Y-axis clearly showed two groups: Pedrinhas, Esteiro do Morro, and Sítio Itapuan are in the negative quadrant, whereas Tapiraí, Eldorado, and Eldorado – Toca da Onça and Sete Barras, and three samples from Esteiro do Morro are in the positive quadrant (Supplementary Material S3). The most suitable K value chosen by the BIC is K = 2. As such, the results of the dPCA analyses clearly show two distinct groups in the X-axis of the first discriminant variable (Figure 4). The AMOVA analyses showed that the variation among individuals (80.70%) was greater than among populations (19.27%) or when considering two groups (lowland × inland/mountain, 17.38%). Mantel’s test suggests isolation by distance (regression coefficient = 0.57, P = 0.003).
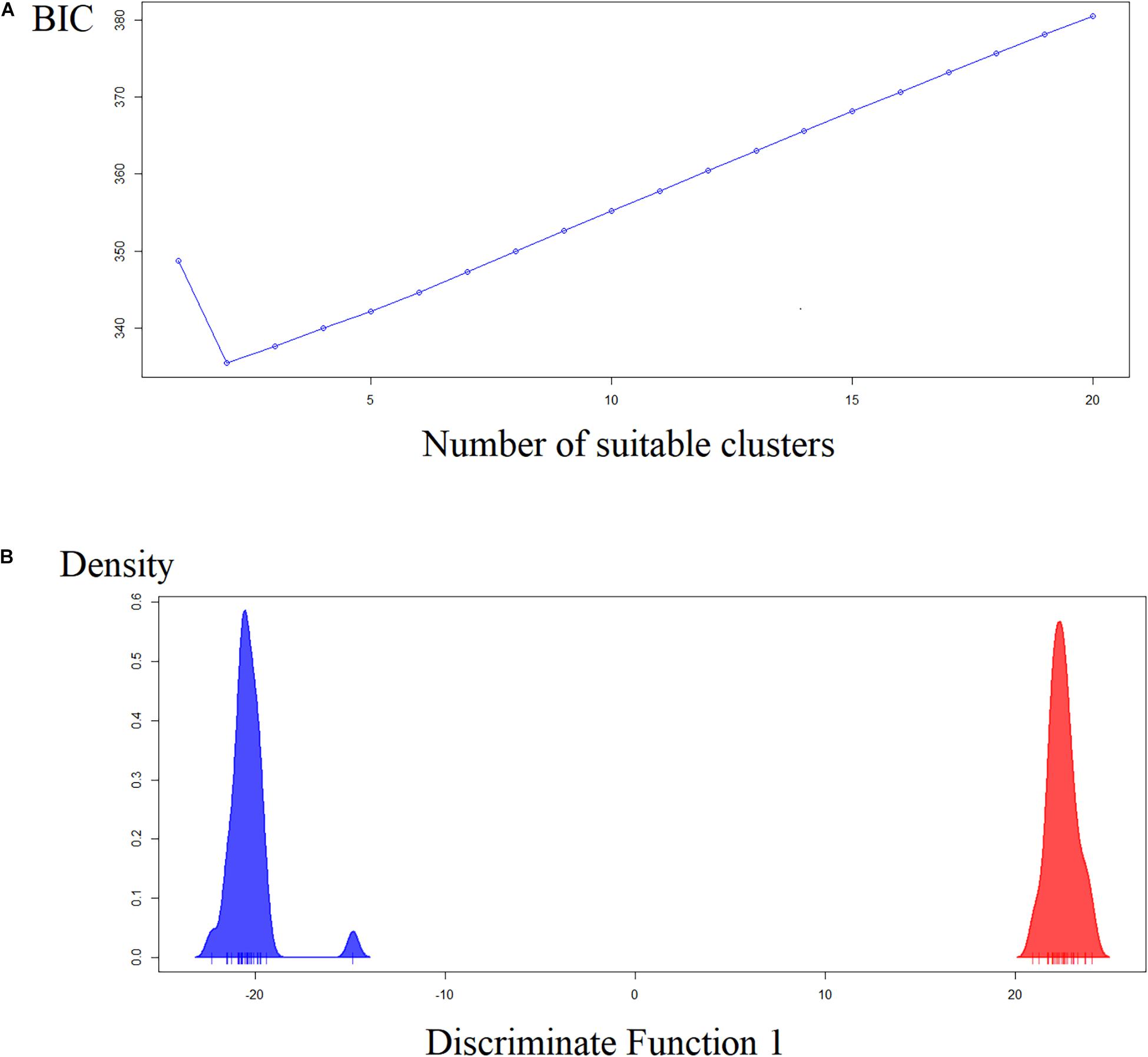
Figure 4. Discriminant analysis of principal components (dPCA). (A) Likely number of clusters estimated by Bayesian Criterion (BIC). (B) Ordination showing the two clusters along the X-axis.
The BayeScan analysis identified 18 outliers. Conversely, the LFMM analysis, which considered the environmental variables, showed that most putative loci under selection were associated with elevation (38 loci) and distance (27 loci). In comparison, 11 and 6 loci were associated with slope and mangrove, and eight were associated with forest and restinga, respectively (Table 4). In contrast, some loci were shared among environmental variables (Table 4). Fourteen loci overlapped in both analyses all associated with distance in the LFFM analysis.
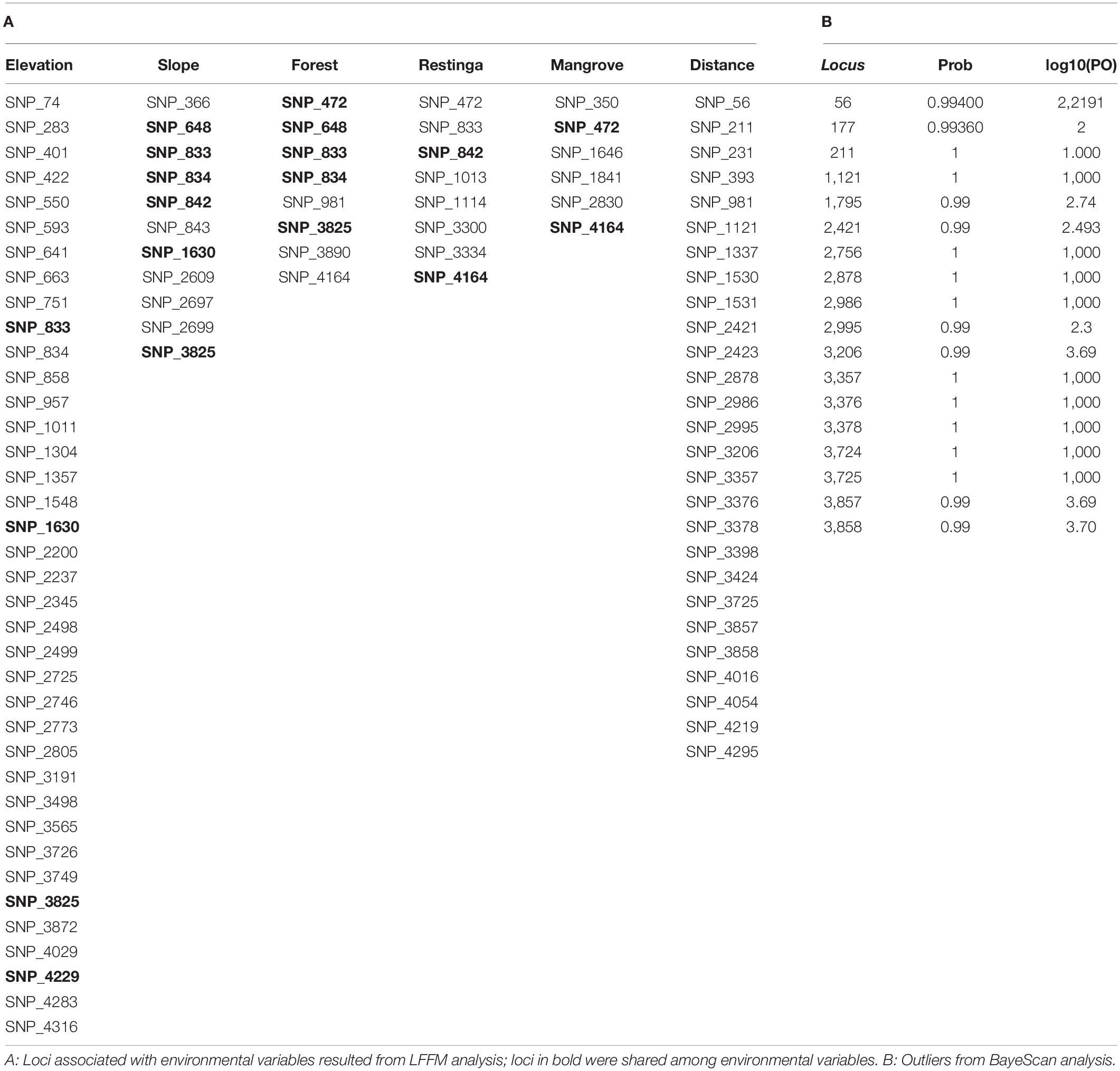
Table 4. Putative loci under adaptive selection.
Discussion
Multiple studies have indicated that the Ke. cruzii may represent a species complex (Ramírez and Dessen, 2000a; Ramírez and Dessen, 2000b; Carvalho-Pinto and Lourenço-de-Oliveira, 2004; Rona et al., 2010; Rona et al., 2013; de Rezende Dias et al., 2018; Kirchgatter et al., 2020). In this study, we used the nextRAD generation sequence approach to investigate the patterns of the genetic structure of Ke. cruzii in the Ribera Valley, South-eastern São Paulo state, Brazil. In addition, we verified the association between the genetic structure and landscape variables. The results of our analyses revealed the presence of two distinct lineages of this species in the studied regions and that they are associated with elevation and isolation by distance. The first lineage corresponds to lowland samples (from Pedrinhas, Sítio Itapuan, and Esteiro do Morro), and the second lineage is composed of specimens from inland and mountain sites (from Sete Barras, Eldorado, and Eldorado Toca da Onça, and Tapiraí). The results of PCA, Structure, and SplitTree analyses showed that in Esteiro do Morro, both lineages coexist in sympatry.
Similar results were found by de Rezende Dias et al. (2018) using sequences from the cpr and clock nuclear genes. Although using a broader geographic area – encompassing three Brazilian states, RJ, SP, and SC – de Rezende Dias et al. (2018) used specimens collected in lowland and inland/mountain localities (Serra do Mar). Their results showed one lineage that corresponds to samples from lowland sites and a second lineage formed of specimens from the Serra do Mar mountain range. Likewise, specimens collected in Bocaina were clustered in both lineages and were found in sympatry. Unlike our results, however, their findings were not associated with isolation by distance, and the Fst values found between the lineages were higher (0.57) than the value found in our study (∼0.25, between lineages). Consequently, de Rezende Dias et al. (2018) strongly suggests the existence of two cryptic species under the name of Ke. cruzii in the study region.
Although the Fst values calculated using NGS datasets generated for samples employed in this study were relatively low, they were statistically significant, except for the values comparing populations from Eldorado and Sete Barras localities. The Fst values obtained from comparisons within lowland and inland and mountain samples were lower (ranging from 0.01 to 0.03 among lowland and 0.001 to 0.038 among inland and mountain samples) than between these regions (in which Fst varied from 0.1 to 0.28), evidence of restricted gene flow between these lineages. Taken together with LFMM analyses (Table 4), genetic differentiation was associated with elevation and distance, despite differences in vegetation type, forest cover, or slope.
Altitudinal stratification in Ke. cruzii was also verified by Lorenz et al. (2014) in the Cananéia municipality when using COI barcode sequences and wing geometric morphometrics. The COI haplotypes were very polymorphic; however, only two of the 60 haplotypes were shared by lowland and hilltop samples. Therefore, considering the results of Lorenz et al. (2014), de Rezende Dias et al. (2018), and our results, we can consider elevational speciation, an ecological speciation in which adaptive divergence leads to dichotomous low/high altitude distribution. Elevational speciation is well known and has been observed in birds, frogs, and plants (Badyaev and Ghalambor, 2001; Caro et al., 2013; Chapman et al., 2013; Funk et al., 2016).
Usually, elevational speciation is studied and observed at high elevations (>1,000 m). However, we observed that small differences among altitudes (∼50 m) showed local adaptation in Ke. cruzii. Lowlands were also associated with high species richness and abundance in Culicidae, which uses bromeliads as larval habitats in Cananéia, southeast Brazil (Marques et al., 2012). Accordingly, even small variations, such as 200 m in elevation, can imply differences in the structure of Culicidae bromeliad communities. Similarly, bacterial and eukaryotic communities of phytotelma, on which the larval stages develop, also vary at low and high elevations (Gilbert et al., 2020; Malfatti et al., 2020). Additionally, Culicidae species distribution varies according to bromeliad species; for example, Culex (Microculex) spp. are more commonly found in Vriesea friburgensis than in Aechmea lindenii, whereas Wyeomyia incaudata and Wy. pilicauda are primarily associated with A. lindenii (Müller and Marcondes, 2006).
Furthermore, altitude is an important factor for species distribution in the bromeliads of the Atlantic Rain Forest (Brandão et al., 2009; Fontoura et al., 2012). For example, Aechmea catendensis and Aechmea serragrandensis were found clustered in lowlands, while Echinocactus sessiliflorus and Aechmea guainumbiorum were found mainly in the Submontana region in South and Southeast Brazil. Conversely, Aechmea cephaloides is typical of the highlands (Fontoura et al., 2012). A similar pattern occurs within Vriesea, the most common genus of Brazilian bromeliad, with species distribution differing at distinct altitudinal ranges (Malfatti et al., 2020). Considering (1) the evolutionary relationship between Ke. cruzii and bromeliads, (2) the evidence that bromeliad species follow a pattern of elevational distribution, and (3) that culicids are distributed according to bromeliad species, we can hypothesize that the lineages found herein may be associated with different bromeliad species and their elevational distribution pattern.
Although our results showed two distinct lineages in the Ribeira Valley, São Paulo, Brazil, Fst values did not corroborate the existence of cryptic or sister species under Ke. cruzii in this region. The differences between these lineages were mainly associated with isolation by distance and elevation, more than the vegetation mosaic, slope, or forest cover. Therefore, we conclude that this species may be under elevational speciation in this region and hypothesize that it is likely associated with the distribution of bromeliad species. In order to confirm our hypothesis, further investigations need to be performed in the region, to verify potential association between bromeliads species and population structure in K. cruzii.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: NCBI SRA BioProject, accession no: PRJNA729988, https://www.ncbi.nlm.nih.gov/bioproject/PRJNA729988.
Author Contributions
BD-S and MAMS conceived the study. BD-S conducted the field collection work, performed the NGS and population analyses, and wrote the manuscript. GL contributed with the environmental metrics and analyses. TO helped with laboratory and NGS analyses. All authors read and agreed with the final version of this manuscript.
Funding
This study was supported by the Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP grants no. 2016/08551-4 to BD-S; no. 2014/26229-7 to MAMS; and no. 2014/09774-1 to GL), and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq grant no. 302375/2020-1 to MAMS and 307432/2019-0 to GL).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors are grateful to the Fundação de Amparo à Pesquisa do Estado de São Paulo and Conselho Nacional de Desenvolvimento Científico e Tecnológico for financial support.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2021.707642/full#supplementary-material
Footnotes
References
Badyaev, A. V., and Ghalambor, C. K. (2001). Evolution of life histories along elevational gradients: trade-off between parental care and fecundity. Ecology 82, 2948–2960. doi: 10.1890/0012-9658(2001)082[2948:eolhae]2.0.co;2
Bourke, B. P., Conn, J. E., Oliveira, T. M. P., Chaves, L. S. M., Bergo, E. S., Laporta, G. Z., et al. (2018). Exploring malaria vector diversity on the Amazon frontier. Malar. J. 17:342. doi: 10.1186/s12936-018-2483-2
Brandão, S. N., Silva, W. N., Silva, L. A. E., Fagundes, V., De Mello, C. E. R., Zimbrão, G., et al. (2009). “Analysis and visualization of the geographical distribution of atlantic forest bromeliads species,” in Proceedings of the IEEE, Symposium on Computational Intelligence and Data Mining, (Nashville: IEEE), 375–380. doi: 10.1109/CIDM.2009.4938674
Branquinho, M. S., Marrelli, M. T., Curado, I., Natal, D., Barata, J. M. S., Tubaki, R., et al. (1997). Infecção do Anopheles (Kerteszia) cruzii por Plasmodium vivax e Plasmodium vivax variante VK247 nos municípios de São Vicente e Juquitiba, São Paulo. Rev. Panam. Salud. Publica 2, 122–134.
Brasil, P., Zalis, M. G., de Pina-Costa, A., Siqueira, A. M., Júnior, C. B., Silva, S., et al. (2017). Outbreak of human malaria caused by Plasmodium simium in the Atlantic Forest in Rio de Janeiro: a molecular epidemiological investigation. Lancet Glob. Health 5, e1038–e1046.
Buery, J. C., Rezende, H. R., Natal, L., Silva, L. S. D., Menezes, R. M. T., Fux, B., et al. (2018). Ecological characterization and infection of Anophelines (Diptera: Culicidae) of the Atlantic Forest in the southeast of Brazil over a 10 year period: has the behavior of the autochthonous malaria vector changed? Mem. Inst. Oswaldo Cruz. 113, 111–118. doi: 10.1590/0074-02760170225
Caro, L. M., Caycedo-Rosales, P. C., Bowie, R. C. K., Slabbekoorn, H., and Cadena, C. D. (2013). Ecological speciation along an elevational gradient in a tropical passerine bird? J. Evol. Biol. 26, 357–374. doi: 10.1111/jeb.12055
Carvalho-Pinto, C. J., and Lourenço-de-Oliveira, R. (2004). Isoenzymatic analysis of four Anopheles (Kerteszia) cruzii (Díptera: Culicidae) populations of Brazil. Mem. Inst. Oswaldo Cruz. 99, 471–475. doi: 10.1590/S0074-02762003000800012
Chapman, M. A., Hiscock, S. J., and Filatov, D. A. (2013). Genomic divergence during speciation driven by adaptation to altitude. Mol. Biol. Evol. 30, 2553–2567. doi: 10.1093/molbev/mst168
Chhatre, V. E., and Emerson, K. J. (2017). StrAuto: automation and parallelization of structure analysis. BMC Bioinformatics 18:192. doi: 10.1186/s12859-017-1593-0
Consoli, R. A. G. B., and Lourenço-de-Oliveira, R. (1994). R: Principais Mosquitos de Importância Sanitária no Brasil. Rio de Janeiro: Brasil Press, 97–114.
Dalla Bona, A. C., and Navarro-Silva, M. A. (2010). Physiological age and longevity of Anopheles (Kerteszia) cruzii Dyar & Knab (Diptera: Culicidae) in the Atlantic Forest of Southern Brazil. Neotrop. Entomol. 39, 282–288. doi: 10.1590/S1519-566X2010000200021
Danecek, P., Auton, A., Abecasis, G., Albers, C. A., Banks, E., DePristo, M. A., et al. (2011). The variant call format and VCFtools. Bioinformatics 27, 2156–2158. doi: 10.1093/bioinformatics/btr330
de Alvarenga, D. A. M., Culleton, R., de Pina-Costa, A., Rodrigues, D. F., Bianco, C. Jr., Silva, S., et al. (2018). An assay for the identification of Plasmodium simium infection for diagnosis of zoonotic malaria in the Brazilian Atlantic Forest. Sci. Rep. 8:86. doi: 10.1038/s41598-017-18216-x
de Rezende Dias, G., Fujii, T. T. S., Fogel, B. F., Lourenço-de-Oliveira, R., Silva-do-Nascimento, T. F., Pitaluga, A. N., et al. (2018). Cryptic diversity in an Atlantic Forest malaria vector from the mountains of South-East Brazil. Parasit. Vectors 15:36. doi: 10.1186/s13071-018-2615-0
Deane, L. M., Ferreira Neto, J. A., Deane, M. P., and Silveira, I. P. S. (1970). Anopheles (Kerteszia) cruzii, a natural vector of the monkey malaria parasites, Plasmodium simium and Plasmodium brasilianum. Trans. R. Soc. Med. Hyg. 64:647. doi: 10.1016/0035-9203(70)90088-x
Demari-Silva, B., Laporta, G. Z., Oliveira, T., and Sallum, M. (2020). Plasmodium infection in Kerteszia cruzii (Diptera: Culicidae) in the Atlantic tropical rain forest, southeastern Brazil. Infect. Genet. Evol. 78:104061. doi: 10.1016/j.meegid.2019.104061
Duarte, A. M. R., Pereira, D. M., de Paula, M. B., Fernandes, A., Urbinatti, P. R., Ribeiro, A. F., et al. (2013). Natural infection in anopheline species and its implications for autochthonous malaria in the Atlantic forest in Brazil. Parasit. Vectors 6:58. doi: 10.1186/1756-3305-6-58
Earl, D. A., and vonHoldt, B. M. (2012). Structure harvester: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Cons. Gen. Resour. 4, 359–361. doi: 10.1007/s12686-011-9548-7
Evanno, G., Regnaut, S., and Goudet, J. (2005). Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol. 14, 2611–2620. doi: 10.1111/j.1365-294X.2005.02553.x
Excoffier, L., and Lischer, H. E. L. (2010). Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 10, 564–567. doi: 10.1111/j.1755-0998.2010.02847
Foll, M., and Gaggiotti, O. E. (2008). A genome scan method to identify selected loci appropriate for both dominant and codominant markers: a bayesian perspective. Genetics 180, 977–993. doi: 10.1534/genetics.108.092221
Fontoura, T., Scudeller, V. V., and da Costa, A. F. (2012). Floristics and environmental factors determining the geographic distribution of epiphytic bromeliads in the Brazilian Atlantic rain forest. Flora 207, 662–672. doi: 10.1016/j.flora.2012.05.003
Forattini, O. P. (2002). Culicidologia Médica. São Paulo: Editora da Universidade de São Paulo Press.
Forattini, O. P., Gomes, A. C., Natal, D., and Santos, J. L. F. (1986). Observações sobre a atividade de mosquitos Culicidae em matas primitivas da planície e perfis epidemiológicos de vários ambientes do Vale do Ribeira, SãoPaulo, Brasil. Rev. Saude Publ. 20, 178–203. doi: 10.1590/S0034-89101986000300002
Foster, P. G., de Oliveira, T. M. P., Bergo, E. S., Conn, J. E., Sant’Ana, D. C., Nagaki, S. S., et al. (2017). Phylogeny of Anophelinae using mitochondrial protein coding genes. R. Soc. Open. Sci. 4:170758. doi: 10.1098/rsos.170758
Frichot, E., Schoville, S. D., Bouchard, G., and François, O. (2013). Testing for associations between loci and environmental gradients using latent factor mixed models. Mol. Biol. Evol. 30, 1687–1699. doi: 10.1093/molbev/mst063
Funk, W. C., Murphy, M. A., Hoke, K. L., Muths, E., Amburgey, S. M., Lemmon, E. M., et al. (2016). Elevational speciation in action? Restricted gene flow associated with adaptive divergence across an altitudinal gradient. J. Evol. Biol. 29, 241–252. doi: 10.1111/jeb.12760
Gilbert, K. J., Bittleston, L. S., Naïve, M. A. K., Kiszewski, A. E., Buenavente, P. A. C., Lohman, D. J., et al. (2020). Investigation of an elevational gradient reveals strong differences between bacterial and eukaryotic communities coinhabiting nepenthes phytotelmata. Microb. Ecol. 80, 334–349. doi: 10.1007/s00248-020-01503-y
Huson, D. H., and Bryant, D. (2006). Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 3, 254–267. doi: 10.1093/molbev/msj030
Jakobsson, M., and Rosenberg, N. A. (2007). CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23, 1801–1806. doi: 10.1093/bioinformatics/btm233
Jombart, T. (2008). Adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24, 1403–1405. doi: 10.1093/bioinformatics/btn129
Jombart, T., and Ahmed, I. (2011). Adegenet 1.3-1: new tools for the analysis of genome-wide SNP data. Bioinformatics 27, 3070–3071. doi: 10.1093/bioinformatics/btr521
Kirchgatter, K., Guimarães, L. O., Trujillano, H. H. Y., Arias, R. F., Cáceres, A. G., Duarte, A. M. C., et al. (2020). Phylogeny of Anopheles (Kerteszia) (Diptera: Culicidae) using mitochondrial genes. Insects 24:324. doi: 10.3390/insects11050324
Kirchgatter, K., Tubaki, R. M., dos Santos Malafronte, R., Alves, I. C., Lima, G. F., Guimarães, L. D. O., et al. (2014). Anopheles (Kerteszia) cruzii (Diptera: Culicidae) in peridomiciliary area during asymptomatic malaria transmission in the Atlantic Forest: molecular identification of blood-meal sources indicates humans as primary intermediate hosts. Rev. Inst. Med. Trop. Sao Paulo. 56, 403–409. doi: 10.1590/S0036-46652014000500006
Kopelman, N. M., Mayzel, J., Jakobsson, M., Rosenberg, N. A., and Mayrose, I. (2015). Clumpak: a program for identifying clustering modes and packaging population structure inferences across K. Mol. Ecol. Resour. 15, 1179–1191. doi: 10.1111/1755-0998.12387
Laporta, G. Z., Burattini, M. N., Levy, D., Fukuya, L. A., de Oliveira, T. M., Maselli, L. M., et al. (2015). Plasmodium falciparum in the southeastern Atlantic forest: a challenge to the bromeliad-malaria paradigm? Malar. J. 25:181. doi: 10.1186/s12936-015-0680-9
Lischer, H. E. L., and Excoffier, L. (2012). PGDSpider: an automated data conversion tool for connecting population genetics and genomics programs. Bioinformatics 28, 298–299. doi: 10.1093/bioinformatics/btr642
Lorenz, C., Marques, T. C., Sallum, M. A. M., and Suesdek, L. (2014). Altitudinal population STRUCTURE and microevolution of the malaria vector Anopheles cruzii (Diptera: Culicidae). Parasit Vectors 16:581. doi: 10.1186/s13071-014-0581-8
Malfatti, E., Ferreira, P., and Utz, L. R. P. (2020). Eukaryotic communities in bromeliad phytotelmata: how do they respond to altitudinal diferences? Diversity 12:326. doi: 10.3390/d12090326
Marques, T. C., Bourke, B. P., Laporta, G. Z., and Sallum, M. A. M. (2012). Mosquito (Diptera: Culicidae) assemblages associated with Nidularium and Vriesea bromeliads in Serra do Mar, Atlantic Forest, Brazil. Parasit. Vectors 5:41. doi: 10.1186/1756-3305-5-41
Medeiros-Sousa, A. R., de Oliveira, R. C., Duarte, A. M. R. C., Mucci, L. F., Ceretti-Junior, W., and Marrelli, M. T. (2019). Effects of anthropogenic landscape changes on the abundance and acrodendrophily of Anopheles (Kerteszia) cruzii, the main vector of malaria parasites in the Atlantic Forest in Brazil. Malar. J. 18:110. doi: 10.1186/s12936-019-2744-8
Miller, M. R., Dunham, J. P., Amores, A., Cresko, W. A., and Johnson, E. A. (2007). Rapid and cost-effective polymorphism identification and genotyping using restriction site associated DNA (RAD) markers. Genome Res. 17, 240–248. doi: 10.1101/gr.5681207
Müller, G. A., and Marcondes, C. B. (2006). Bromeliad-associated mosquitoes from Atlantic forest in Santa Catarina Island, southern Brazil (Diptera, Culicidae), with new records for the State of Santa Catarina. Iheringia Ser. Zool. 96, 315–319. doi: 10.1590/S0073-47212006000300007
Oliveira, T. M., Foster, P. G., Bergo, E. S., Nagaki, S. S., Sanabani, S. S., Marinotti, O., et al. (2016). Mitochondrial genomes of Anopheles (Kerteszia) (Diptera: Culicidae) from the Atlantic Forest, Brazil. J. Med. Entomol. 53, 790–797.
Pembleton, L., Cogan, N., and Forster, J. (2013). StAMPP: an R package for calculation of genetic differentiation and structure of mixed-ploidy level populations. Mol. Ecol. Resour. 13, 946–952. doi: 10.1111/1755-0998.12129
Pritchard, J. K., Stephens, P., and Donnelly, P. (2000). Inference of population structure using multilocus genotype data. Genetics 155, 945–959. doi: 10.1093/genetics/155.2.945
Ramírez, C. C., and Dessen, E. M. (2000a). Chromosomal evidence for sibling species of the malaria vector Anopheles cruzii. Genome 43, 143–151. doi: 10.1139/g99-103
Ramírez, C. C., and Dessen, E. M. (2000b). Chromosome differentiated populations of Anopheles cruzii: evidence for a third sibling species. Genetica 108, 73–80. doi: 10.1023/a:1004020904877
Rona, L. D., Carvalho-Pinto, C. J., and Peixoto, A. A. (2010). Molecular evidence for the occurrence of a new sibling species within the Anopheles (Kerteszia) cruzii complex in south-east Brazil. Malar. J. 9:33. doi: 10.1186/1475-2875-9-33
Rona, L. D., Carvalho-Pinto, C. J., and Peixoto, A. A. (2013). Evidence for the occurrence of two sympatric sibling species within the Anopheles (Kerteszia) cruzii complex in southeast Brazil and the detection of asymmetric introgression between them using a multilocus analysis. BMC Evol. Biol. 13:207. doi: 10.1186/1471-2148-13-207
Rosenberg, N. A. (2004). Distruct: a program for the graphical display of population structure. Mol. Ecol. Notes 4, 137–138. doi: 10.1046/j.1471-8286.2003.00566.x
Russello, M. A., Waterhouse, M. D., Etter, P. D., and Johnson, E. A. (2015). From promise to practice: pairing non-invasive sampling with genomics in conservation. PeerJ. 3:e1106. doi: 10.7717/peerj.1106
Keywords: Kerteszia cruzii, population structure, landscape, isolation by distance, elevational speciation
Citation: Demari-Silva B, Laporta GZ, de Oliveira TMP and Sallum MAM (2021) Evidence of Elevational Speciation in Kerteszia cruzii (Diptera: Culicidae) in the Ribeira Valley, São Paulo, Brazil. Front. Ecol. Evol. 9:707642. doi: 10.3389/fevo.2021.707642
Received: 10 May 2021; Accepted: 01 June 2021;
Published: 06 July 2021.
Edited by:
Kaio Cesar Chaboli Alevi, São Paulo State Universty, BrazilReviewed by:
Hélcio Reinaldo Gil-Santana, Oswaldo Cruz Foundation (Fiocruz), BrazilCarlos José Carvalho Pinto, Federal University of Santa Catarina, Brazil
Copyright © 2021 Demari-Silva, Laporta, de Oliveira and Sallum. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bruna Demari-Silva, bdemari.silva@gmail.com